
Selective micellar electrokinetic chromatographic method for simultaneous determination of some pharmaceutical binary mixtures containing non-steroidal anti-inflammatory drugs
2013-12-23 06:15MichaelElKommosNiveenMohamedAhmedAbdelHakiem
Michael E. El-Kommos, Niveen A. Mohamed, Ahmed F. Abdel Hakiem
Department of Pharmaceutical Analytical Chemistry, Faculty of Pharmacy, Assiut University, Assiut 71526, Egypt
1. Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) are widely used as analgesics and at higher doses, as anti-inflammatory in the treatment of rheumatic diseases and other musculoskeletal disorders. They are also available as over-the counter pharmaceutical preparations. The safety and efficacy of these drugs are critically related to whether or not their contents conform to labeled amounts [1,2]. They are introduced all over the world in many pharmaceutical formulations either alone or with adjuvant drugs whether to synergize, or to impart added effect.
So, an urgent need arises for the development of analytical methods for their determination in their pharmaceutical mixtures. Five commonly prescribed pharmaceutical binary mixtures containing three NSAIDs were selected for our analytical study. Literature survey revealed that these selected binary mixtures were analyzed by high performance liquid chromatography (HPLC) [3-11] in addition to UV-chemometric assisted spectrophotometric methods [12-14]. Capillary electrophoresis (CE) has been used for the determination of NSAIDs [2,15-19]. However, the analysis of binary mixtures of these drugs with muscle relaxants, local anesthetics and analgesics by CE was not found in the literature. Micellar electrokinetic chromatography (MEKC) is a hybrid of electrophoresis and chromatography introduced by terabe in 1984. MEKC is one of the most widely used CE modes.MEKC is characterized by that it can be used for the separation of neutral solutes as well as charged ones. The separation of neutral species is accomplished by the use of surfactant in the running buffer in concentration above the critical micelle concentration. The neutral solutes arranged themselves in and out of the micelles and move according to the micelle velocity not by their electrophoretic mobility resulting in good separation of the mixture [20].
The chemical structures of the drugs contained in the investigated binary mixtures are shown in Fig.1. In the present work, a very simple MEKC technique was applied for the first time for the separation and simultaneous determination of seven drugs in a synthetic mixture and then applied to the determination of the drugs in five pharmaceutical binary mixtures. The studied drugs are three NSAIDs either with anesthetic,analgesic antipyretic or skeletal muscle relaxants or other analgesics. The investigated mixtures were IP-PC, IPCZ, IP-MC, KP-CZ and DS-LC. The literature survey revealed that there is no reported CE method in the literature for the separation and simultaneous determination of the investigated binary mixtures.The aim of the present work was to develop an MEKC method for the analysis of the active constituents of these commercially available mixtures under the same conditions and to offer the possibility of analyzing possible future binary mixtures containing NSAIDs with skeletal muscle relaxants, analgesic, or local anesthetic.
2. Experimental
2.1. Apparatus and electrophoretic conditions
A CE system (G 1600A model, Agilent technologies,Waldbronn, Germany) with UV diode-array detector (DAD)was used. It consists of fused silica (Polymicro Technologies,Phoenix, AZ, USA) of 65 cm total length with 50 μm internal diameter. The capillary temperature was set at 25°C. The separation was performed using borate buffer (20 mM, pH 9)containing 15% (v/v) methanol and 100 mM SDS. The running buffer was filtered through 0.45 μm pore size membrane filter of 30 mm in diameter. Hydrodynamic injection of the sample vial was applied with the aid of pressure mode for 10 S at 50 mbar. Diode-array detector was set at 214 nm.Before start-up, the capillary was preconditioned with varying concentrations of NaOH on different periods of time at room temprature and finally with running buffer in order to activate the interior wall of the capillary. Between runs the capillary was rinsed with running buffer for 5 min. Ultrasonic cleaner(Cole-Parmer, Chicago, USA), pH meter, model 3305 (Jenway, London, UK) and sartorius handy balance—H51 (Hanover, Germany) were used throughout the experiments.
2.2. Materials and reagents

Fig.1 Structures of the investigated drugs.
Pharmaceutical grade CZ (% purity 98.10±0.86) and PC(%purity 98.76±1.85) were obtained from GlaxoSmithKline Co. (Cairo, Egypt), DS (% purity 96.42±1.78), IP (% purity 97.23±0.92), KP (% purity 98.76±1.82) and LC (% purity 99.7±1.25)were from Pharco Pharmaceuticals Co.(Alexandria,Egypt),MC(%purity 97.05±1.12)was from Eva Pharma for Pharmaceuticals & Medical Appliances S.A.E Co. (Cairo,Egypt). All investigated drugs were subjected to official methods [21,22] to determine their purity.
Pharmaceutical preparations containing the studied drugs were purchased from the local market.
Cetafen tab. IP (200) and PC (325) were obtained from Queen Pharma International, Cairo, Egypt, Flexofan cap. KP(50) and CZ (250) from Amirya Pharma. Ind., Alexandria,Egypt, Ibuflex tab. IP (400) and MC (750) from GlobalNap Pharmaceutical 6 October, Giza, Egypt, Myofen cap. IP (200)and CZ(250)from Eva Pharma,Cairo,Egypt,Olfen amp.DS(75) and LC (25) from MUP, Abusultan, Ismailia, Egypt.
SDS was obtained from Sigma-Aldrich, St Louis, USA.
Ultrapure Milli-Q water (Millipore, Bedford, MA, USA)was used for preparation of running buffers. All chemicals used for the preparation of buffer electrolytes were of analytical- reagent grade.
2.3. Preparation of working standard solutions
Accurately weighed 6, 12, 30, 50, 50 and 110 mg of IP, CZ, KP,DS, LC or MC, respectively were transferred to 10 mL volumetric flasks containing 5 mL methanol. The contents of the flasks were shaken well and completed to the mark with methanol to obtain the stock standard solutions of the studied drugs.Further dilutions of the stock solutions with methanol were made to get working standard solutions in the following concentration ranges: 0.06-1.8 mg/mL for CZ, 0.27-4.8 mg/mL for DS, 0.03-0.8 mg/mL for IP, 0.20-2.2 mg/mL for KP, 0.30-4.4 mg/mL for LC, 1.10-10.1 mg/mL for MC and 0.03-1.0 mg/mL for PC.
2.4. Preparation of synthetic mixture solution
Into 10 mL volumetric flask,known accurate volumes of stock standard solutions of the seven investigated drugs were mixed and diluted to contain 0.5 mg/mL of CZ, 4.2 mg/mL of DS,non-steroidal anti-inflammatory drugs (NSAIDs), 0.4 mg/mL of IP,0.3 mg/mL of KP,1.4 mg/mL of LC,1.5 mg/mL of MC and 0.64 mg/mL of PC.
2.5. Analysis of dosage forms
2.5.1. Capsules
The contents of 20 Flexofan capsules were accurately weighed,evacuated, finely powdered and mixed thoroughly. An accurately weighed amount of mixed powder equivalent to 0.1 mg KP and 0.5 mg CZ was transferred to 10 mL calibrated flask,dissolved in about 7 mL methanol (high performance liquid chromatography grade), sonicated for 10 min, diluted to the mark with methanol, mixed well and then filtered. The first portion of the filtrate was rejected. The prepared solution was diluted quantitatively to obtain the required concentration for the assay.
2.5.2. Tablets
Twenty tablets were accurately weighed, finely powdered and mixed thoroughly. Known amount of powdered Cetafen tablets®mix equivalent to 0.15 mg IP and 0.24 mg PC was accurately weighed and treated as capsules to get working sample solution containing 18.3 μg/mL IP and 29.7 μg/mL PC after further dilutions. For Ibuflex tablets®the same previous steps were carried out to get working sample solution containing 18.3 μg/mL IP and 34.3 μg/mL MC.
2.5.3. Ampoules
The contents of ten olfen ampoules®were mixed well and accurately measured volume equivalent to 0.1 mg LC and 0.3 mg DS was transferred to 10 mL calibrated flask, diluted to the mark with methanol ( high performance liquid chromatography grade)and further dilutions were done to get working sample solution containing 45 μg/mL DS and 15 μg/mL LC.
3. Results and discussion
In the present work, the seven drugs were separated and determined by CE(MEKC mode)simultaneously in a laboratory prepared synthetic mixture and the same method was applied for separation and determination of five pharmaceutical binary mixtures. Three of the drugs belong to NSAIDs (DS, IP and KP),formulated with four adjuvants(CZ,LC,MC and PC)and marketed in five commonly prescribed pharmaceutical dosage forms.To our knowledge,no CE method has been developed for the determination of these binary mixtures.The method depends on the difference in polarity of the investigated compounds leading to different electrophoretic mobilities and accordingly different migration times [20].
3.1. Optimization of capillary electrophoretic conditions
All the parameters affecting the efficiency of separation through its effect on electro-osmotic flow were studied and optimized including buffer system composition (type, pH and ionic strength), effects of SDS addition and organic modifier addition as well as analytical voltage.
3.1.1. Buffer system
3.1.1.1. Buffer type. Since the studied drugs are mostly acidic or neutral, the alkaline pH is preferred for their separation because the analysis time is relatively shorter than that obtained at acidic pH, due to the higher electro-osmotic flow(EOF).In addition at alkaline pH,acidic analytes will migrate after the EOF.So phosphate and borate buffers were tested at pH 8. Upon testing the effect of both phosphate and borate buffers on resolution of the studied drugs, better separation efficiency was obtained with borate buffer.These results agree well with most CE separations in the literature for analysis of NSAIDs since they also used borate buffer [17,23].
3.1.1.2. Borate buffer pH and its ionic strength. Borate buffer solutions ranging from pH 8 to pH 9.5 were investigated.The pH of the back ground electrolyte(BGE)is known to play an important role in CE which requires charged analytes,since it contributes to the degree of dissociation of weak acids or bases, thus influencing their effective mobilities [24]. DS, IP and KP have low pKavalues,so they require deprotonation in alkaline medium to be negatively charged. Varying degrees of partition of all analytes from SDS micelles at different pH values interpreted the difference in resolution efficiency. It was found that at pH 8 and pH 8.5 most of the peaks were not resolved and best resolutions were obtained at both pH 9 and 9.5.However,pH 9 was preferred due to shorter analysis time as illustrated in Fig.2. Also, the elution order varied at different pHs due to difference in polarities of the investigated drugs.
Different borate buffer concentrations (5, 10, 20, 40, 60 and 80 mM) were studied at the optimum pH value (pH 9). It is known that increasing the buffer ionic strength modulates the EOF and electrophoretic mobility of the analyte.Additionally,higher concentrations of buffer salts can modulate the effective charge at the capillary wall and consequently modulate the interactions between the wall and the solute [24]. It was found that no separation was achieved upon using 5 or 10 mM borate buffer and the resolution efficiency was gradually increased with increasing the ionic strength till obtaining the best resolution at 20 mM concentration then the resolution started to decrease at higher buffer concentrations. Therefore,the optimum composition of the buffer was 20 mM ionic strength of borate buffer at pH 9.0.
3.1.1.3. Surfactant addition. The effect of addition of SDS(the most commonly used anionic surfactant) to the BGE was studied. This surfactant is characterized by its high aqueous solubility, low critical micellar concentration (CMC) (8.1 mM),small ultraviolet light molar absorptivity, availability and low cost [25]. It was found that addition of SDS gives great improvement for both resolution and peak shapes of all analytes.

Fig.2 MEKC separation of (1) Paracetamol (PC), (2) Methocarbamol (MC), (3) Ketoprofen (KP), (4) Ibuprofen (IP), (5)Diclofenac sodium (DS), (6) Chlorzoxazone (CZ) and (7) Lidocaine hydrochloride(LC)of concentrations 0.64,1.5,0.3,0.4,4.2,0.5 and 1.4 mg/mL, respectively, at different borate buffer pH values; (A) pH 8.00, (B) pH 8.50, (C) pH 9.00, and (D) pH 9.50.
Since all investigated analytes exhibited anionic and neutral characters in alkaline conditions of borate buffer (pH 9),addition of SDS improved their resolution greatly which supported the previously mentioned facts.
Different concentrations of SDS were tested including 40,60,80, 100 and 120 mM. The best resolution was obtained with 100 mM concentration.
3.1.1.4. Type of organic modifier added and its percentage.
Trials were made to separate the investigated binary mixtures without addition of organic modifier to the BGE but they failed, so methanol and acetonitrile were tested in order to improve the separation and determination efficiency. The organic modifiers affect the distribution of the analytes between aqueous phase and micelles, also it can affect the charge state of ionizable groups. Best results were obtained with methanol and this can be explained by the fact that methanol can decrease the zeta potential and reduce the EOF,so it increases the separation efficiency [2]. The effect of methanol percentage in the BGE was studied in the range 5-20%. Further increase in methanol concentration was avoided since it will prolong the migration time and will broaden the peaks [17] because it decrease EOF. It was found that upon using 15% methanol in the BGE, all studied drugs exhibited best resolution efficiencies as illustrated in Fig.3.

Fig.3 MEKC separation of: (1) paracetamol (PC), (2) Methocarbamol (MC), (3) Ketoprofen (KP), (4) Ibuprofen (IP), (5)Diclofenac sodium (DS), (6) Chlorzoxazone (CZ) and (7) Lidocaine hydrochloride(LC)of concentrations 0.64,1.5,0.3,0.4,4.2,0.5 and 1.4 mg/mL, respectively, using different methanol concentrations in the running electrolyte;(A)5%,(B)10%,(C)15%,and (D) 20%.
It is apparent from the study that 10% and 15% methanol in the BGE exhibited the best results. However, when 10%methanol was used, shorter analysis time was obtained and overlapping between IP and KP occurred. On the other hand, when using 15% methanol in the BGE all the investigated drugs were well resolved with symmetric and sharp signals. Therefore, it was selected in the general assay procedure.
3.1.2. Analytical voltage
Analyses at different potentials (5, 10, 15, 20 and 20 kV) were carried out to determine the optimum voltage. Upon trying 5 and 10 kV, we got very bad resolution due to overlapped broad bands for most analytes, while at 15 kV we got the best resolution. Upon raising the applied voltage to 20 kV, overlapping between KP and MC signals occurred.We did not test voltages higher than 20 kV to avoid generation of joule heat inside the capillary which adversely affects the peak shapes and deteriorates the inside capillary wall. However, application of 20 kV is valuable only in case of DS-LC binary mixture where it greatly decreases the separation time. This can be explained by the increase of EOF at higher voltage[20].
3.2. Suggested explanation for the elution order
It was reported that the majority of drugs are either acidic and/or basic water-soluble compounds.The bases for separation in free solution CE relies upon an exploitation of differences between the analytes' electrophoretic mobilities, which are related to solutes, charge and size. Consequently, the separation of many drugs is possible by free-solution capillary electrophoresis(FSCE). For example, an acidic drug may be analyzed in its anionic form at high pH and basic drugs may be tested at low pH in the cationic form. Zwitterionic drugs (those containing both acidic and basic groups) may be analyzed at either end of the pH range. A mixture of neutral drugs would be unresolved by FSCE. However, ionic, charged micelles can be incorporated into the electrolyte solution to add a partitioning element to the separation. This is the basic idea behind MEKC [26]. The seven studied drugs have different ionization constants affecting their separation behavior, paracetamol and methocarbamol (pKa9.5,14.5, respectively) are zweitter ions and their acidic protons are ionized in the alkaline medium of borate buffer(pH 9) resulting in imparting strong negative charge on their molecules undergoing strong repulsion with the negatively charged surface of SDS resulting in their first elution, paracetamol firstly owing to its smaller molecular weight (151.71 g/mole) then methocarbamol of the higher molecular weight (241.2 g/mole), followed by the three NSAIDs acidic drugs which appeared in the following sequence;the most acidic one ketoprofen(pKa4),ibuprofen(pKa4.4) then diclofenac sodium (pKa4.1) which has the highest molecular weight of the three (318.1 g/mole) interpreting its late appearance after ibuprofen, then coming the zweitter ion chlorzoxazone (pKa8.3) which is not greatly affected (weak ionization for its hydroxyl group) at this alkaline pH owing to the presence of strong alkaline nucleus (oxazole ring) keeping a weak basic character for the molecule, finally coming the basic drug lidocaine hydrochloride(pKa7.9 and M.Wt.251.3 g/mole).
3.3. Analytical method validation
Method validation is the process of making analytical technique performance adequate for its intended use in the future[27]. A developed analytical method must ensure a minimum package of validation experiments that are conducted giving satisfactory results. For an acceptable analytical methodology and method development, certain method validation steps are required according to USP 31 NF 26 [22] and ICH guidelines[28]. The validation parameters tested included: Linearity,limit of detection (LOD) and limit of quantitation (LOQ),accuracy, precision, robustness and ruggedness.
3.3.1. Linearity
Under the optimum chromatographic conditions,the relationship between peak areas and concentration was linear and ranged from 0.02 to 4.80 mg/mL for all the studied drugs,which permits efficient determination of them in their pharmaceutical dosage forms especially they have high therapeutic doses. The intercepts (a), slopes (b), correlation coefficients,LOD and LOQ are summarized in Table 1.
The LOD for all the studied drugs ranged from 8.2×10-3to 0.32 mg/mL and LOQ ranged from 0.02 to 1.1 mg/mL.These findings indicate very high sensitivity of the proposed method as illustrated in Table 1.
3.3.2. Accuracy
Accuracy was determined by comparing measured concentrations of IP, PC, CZ, MC, KP, DS and LC with the actual values and expressed as percentage. The accuracy was measured and calculated for each of the studied drugs at threeconcentration levels covering the linear range. The results are shown in Table 2 illustrating high accuracy and excellent recovery percentages ranging from 97.57% to 102.80%.

Table 1 Summary for the quantitative parameters and statistical data using the proposed MEKC method for analysis of studied drugs.
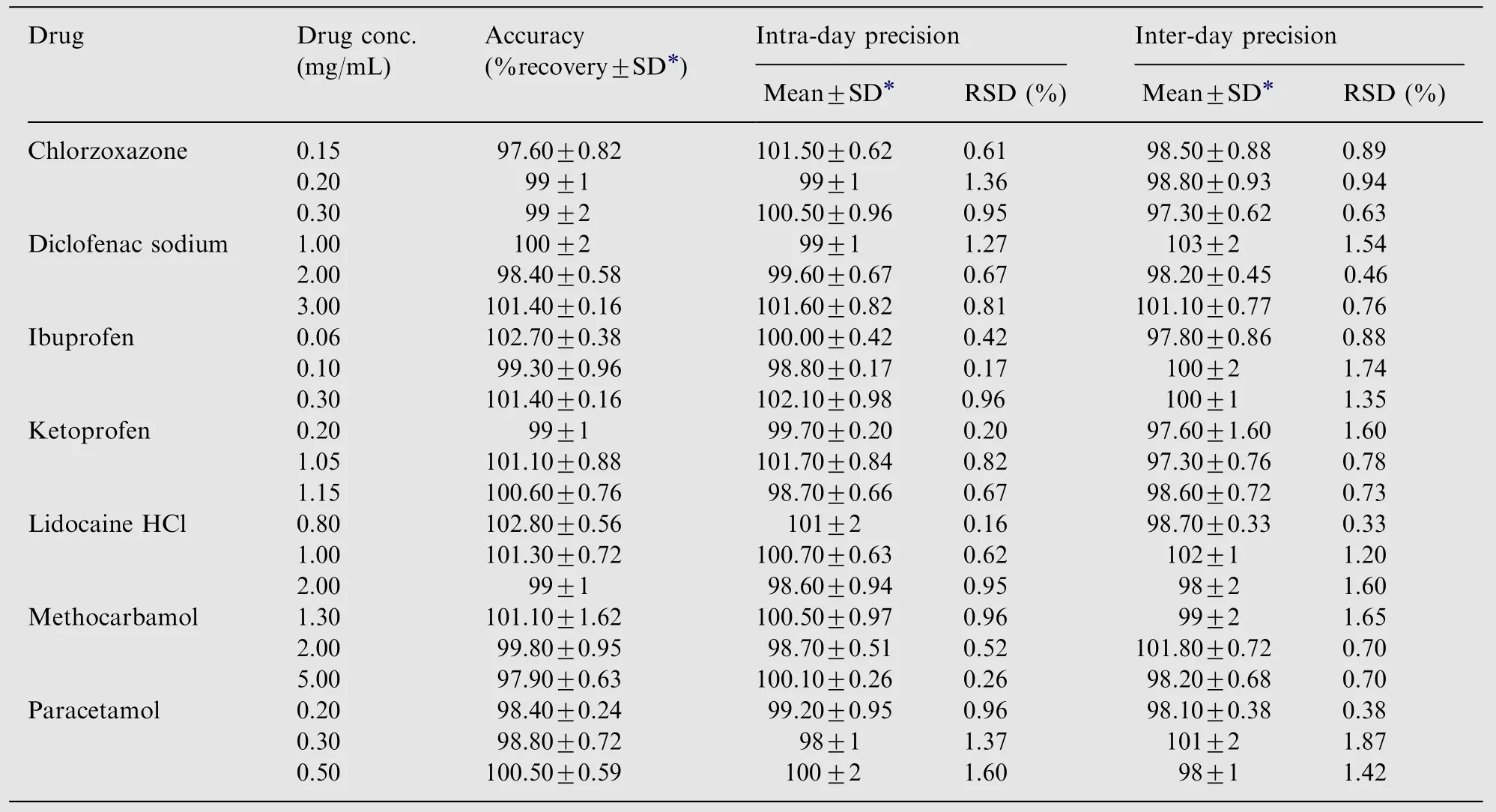
Table 2 Accuracy and precision of the proposed MEKC method.
3.3.3. Precision
Intra-day precision was determined by replicate analysis(n=3) of standard solutions at low (0.15 mg/mL), medium(0.20 mg/mL), and high (0.20 mg/mL) concentration levels Table 2. The inter-day precision was conducted by repeating the analysis over a period of three consecutive days. The overall precision of the method was expressed as relative standard deviations (RSDs). It was found that RSDs for all studied drugs were less than 2% indicating good repeatability and precision.
3.3.4. Robustness
The robustness of an analytical method is defined as a measure of its capacity to remain unaffected by small but deliberate variations in method parameters. The most sensitive electrophoretic parameters that could affect separation performances were examined: buffer pH (±0.2), buffer ionic strength(±0.2 mM), and organic modifier percent (±0.5%). It was found that minor variations of these variables did not significantly affect the performance of the method as shown in Table 3. So the method can be considered robust.
3.3.5. Ruggedness
Ruggedness can be defined as the degree of reproducibility of test results obtained by the analysis of the same samples under a variety of conditions, and it was achieved in our proposed method by its application by different analysts during different days and precise results were obtained. The suggested method was found to be rugged.
Thus, the developed validated MEKC is an accurate,precise, robust and simple method for the determination of five binary mixtures containing some NSAIDs. The method can be used for the analytical quality control of the dosage forms which contain these mixtures.
3.4. Application of the developed MEKC method for pharmaceutical preparations
The proposed method was successfully applied to the assay of the seven drugs in the five pharmaceutical binary mixtures.The proposed method was found to be selective due to cleanness of the electropherogram of pharmaceutical formulations. The average percentage recoveries of different concentrations were based on the average of three replicates determinations and the results obtained were compared with reported HPLC methods by means of t- and F- tests at 95%confidence level for each drug (Table 4). No significant differences were found between the results obtained by the reported HPLC methods and the proposed method for the studied pharmaceutical binary mixtures, indicating good accuracy and precision. Thus the proposed MEKC method can be used in quality control laboratories for simultaneous determination of the investigated binary mixtures.
4. Conclusion
An MEKC method was developed for the simultaneous determination of seven drugs and applied for the separation and determination of five pharmaceutical binary mixtures containing three NSAIDs under the same electrophoretic conditions. The proposed method was validated according to ICH and USP 31 NF 26 guidelines. The validation studyshowed that the method is simple, accurate, precise, selective and economic. The method can be applied in quality control laboratories not only for the investigated binary mixtures but also for possible future binary mixtures containing the same components.

Table 3 Robustness of the proposed MEKC method for analysis of studied drugs (%recoverya±SD).
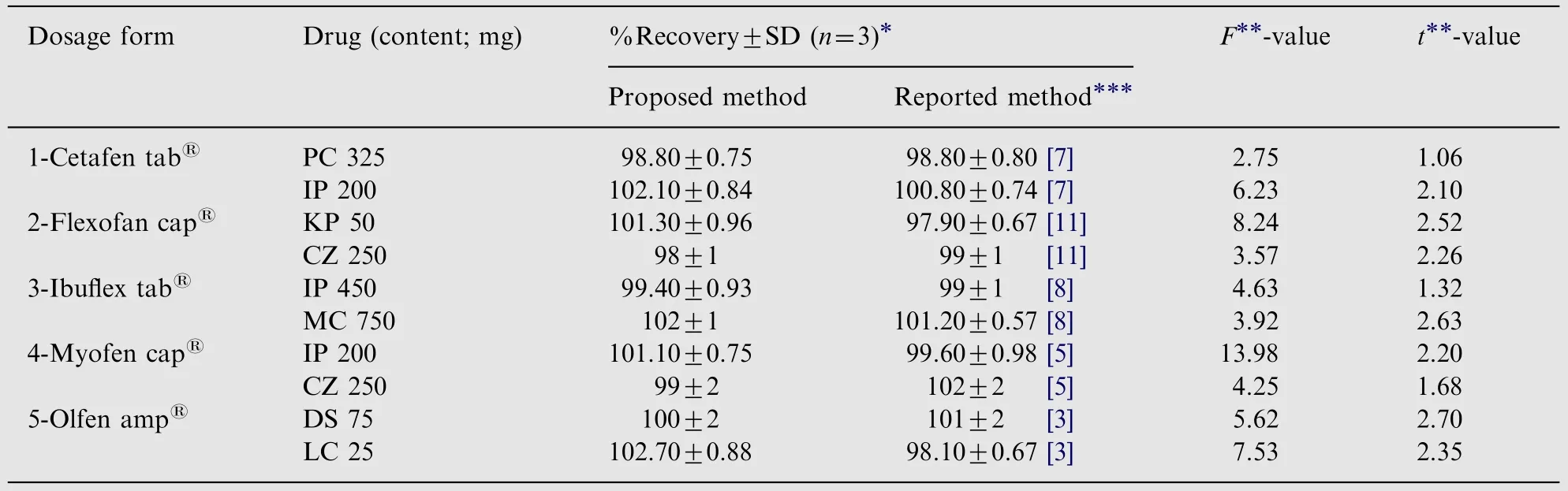
Table 4 Assay of the studied dosage forms using the proposed and reported procedures.
[1] J. N. Delgado, W. A. Remers (Eds.), Wilson and Gisvold's Textbook of Organic Medicinal and Pharmaceutical Chemistry,10th ed., New York, 2004.
[2] Y.L. Chen, S.M. Wu, Capillary zone electrophoresis for simultaneous determination of seven nonsteroidal anti-inflammatory drugs in pharmaceuticals, Anal. Bioanal. Chem. 381 (2005)907-912.
[3] L. Hanysova, M. Mokry, P. Kastner, et al., HPLC evaluation of dichlofenac in the various forms of therapeutic preparations,J. Chem. Pap. 59 (2005) 103-108.
[4] S.T. Hassib, A.A. Mohammad, A.A. El-Zaher, et al., Simultaneous determination of chlorzoxazone and ketoprofen in binary mixtures and in ternary mixtures containing the chlorzoxazone degradation product by reversed-phase liquid chromatography,J. AOAC 90 (2007) 693-699.
[5] S.S. zarapkar, S.S. Kolte, A.A. Dhanvate, et al., High performance liquid chromatographic determination of chloroxazone and ibuprofen, simultaneously from pharmaceutical preparation,Indian Drugs 33 (1996) 275-279.
[6] E.F.Elkady,M.A.Fouad,Two liquid chromatographic methods for the simultaneous determination of ibuprofen and methocarbamol or chloroxazone in the presence their degradation products, J. Liq. Chromatogr. Related Technol. 35 (7) (2012)882-895.
[7] S.V. Erram, S.M. Doshi, V.M. Kulkarni, Simultaneous estimation of ibuprofen and paracetamol in tablets by RP-HPLC., Ind.J. Pharm. Sci. 54 (1992) 122-124.
[8] P.D. Sethi (Ed.), High Performance Liquid Chromatography,Qunatitative Analysis of Pharmaceutical Formulations, vol. 1,CBS Publishers and Distributors, New Delhi-India, 2010 350-389.
[9] I.C. Bhoir, B. Raman, M. Sundaresan, et al., Isocratic simultaneous supercritical fluid liquid chromatography: separation and estimation of Ibuprofen and Methocatbamol in solid dosage form, Indian Drugs 35 (1998) 134-138.
[10] S.S. Zarapkar, U.P. Halkar, N.P. Bhandari, Reverse phase high performance liquid chromatographic determination of Ibuprofen,Paracetamol and Methocatbamol in tablets, Indian Drugs 36(1999) 710-713.
[11] J.W. Jorgenson, K.D. Lukacs, Zone electrophoresis in opentubular galss capillaries, Anal. Chem. 59 (1981) 1298-1302.
[12] W.S. Hassan, Determination of ibuprofen and paracetamol in binary mixtures using chemometric-assisted spectrophotometric methods, Am. J. Appl. Sci. 5 (2008) 1005-1012.
[13] M.R. Khoshayand, H. Abdollahi, M. Shariatpanahi, et al.,Simultaneous spectrophotometric determination of paracetamol,ibuprofen and caffeine in pharmaceuticals by chemometric methods, Spectrochim. Acta, Part A 70 (2008) 491-499.
[14] Y.C. de Micalizzi, N.B. Pappano, N.B. Debattista, First and second order derivative spectrophotometric determination of benzyl alcohol and diclofenac inpharmaceutical forms, Talanta 47 (1998) 525-530.
[16] M.M. Meighana, M. Dawodb, R.M. Guijtb, et al., Pressureassisted electrokinetic supercharging for the enhancement of nonsteroidal anti-inflammatory drugs, J. Chromatogr. A 1218 (2011)6750-6755.
[17] M. Dawod, M.C. Breadmore, R.M. Guijt, et al., Electrokinetic supercharging for on-line preconcentration of seven non-steroidal anti-inflammatory drugs in water samples, J. Chromatogr. A 1189 (2008) 278-284.
[18] A. Macia, F. Borrull, M. Calull, et al., Different sample stacking strategies to analyse some nonsteroidal anti-inflammatory drugs by micellar electrokinetic capillary chromatography in mineral waters, J. Chromatogr. A 1117 (2006) 234-245.
[19] Y.F. Pai, C.Y. Liu, Capillary electrochromatographic separation of non-steroidal anti-inflammatory drugs with a histidine bonded phase, J. Chromatogr. A 982 (2002) 293-301.
[20] D.N. Heiger, An Introduction High Performance Capillary Electrophoresis,second ed., Hewlett-Packard Company,France,1992.
[21] The British Pharmacopoeia, HM Stationary office, London, 2010.
[22] The United States Pharmacopoeia 34 and NF 29, American Pharmaceutical Association, Washington, DC, 2011.
[23] S.Toasaksiri,D.L.Massart,Y.Vander Heyden,Study of method validation criteria in a capillary electrophoresis method for the separation of non-steroidal anti-inflammatory drugs,Anal.Chim.Acta 416 (2000) 29-42.
[24] S. Furlanetto, S. Lanteri, S. Orlandini, et al., Selection of background electrolyte for CZE analysis by a chemometric approach:Part I. Separation of a mixture of acidic non-steroidal antiinflammatory drugs,J.Pharm.Biomed.Anal.43(2007) 1388-1401.
[25] M.G. Khaledi, High Performance Capillary Electrophoresis: Theory, Techniques, and Applications, Wiley, INC., New York, 1998.
[26] K.D.Altria,M.M.Ragon,Introduction to Quantitative Applications of Capillary Electrophoresis in Pharmaceutical Analysis,Beckman Coulter, England, 1993.
[27] I. Ali, H.Y. Aboul-Enein, V.K. Gupta, Precision in capillary electrophoresis, Anal. Lett. 39 (2006) 2345-2357.
[28] Department of Health and Human Services, Food and Drug Administration, Guidance for Industry on Bioanalytical Method Validation; Availability Fed. Regist. 66 (2001) 28526.
 Journal of Pharmaceutical Analysis2013年1期
Journal of Pharmaceutical Analysis2013年1期
- Journal of Pharmaceutical Analysis的其它文章
- On-line coupling of derivatization with pre-concentration to determine trace levels of methotrexate
- Simultaneous determination of atorvastatin, metformin and glimepiride in human plasma by LC-MS/MS and its application to a human pharmacokinetic study
- JPA Prize in 2012
- Study on changes of polyamine levels in mice with the development of U14 cervical cancer
- Quantitation of bivalirudin, a novel anticoagulant peptide,in human plasma by LC-MS/MS: Method development,validation and application to pharmacokinetics
- A rapid and sensitive liquid chromatography-tandem mass spectrometric assay for duloxetine in human plasma: Its pharmacokinetic application