
固相萃取/超高效液相色谱法测定龙胆泻肝丸中栀子苷、龙胆苦苷与黄芩苷
2013-11-28 01:00:08薛昆鹏陈再洁赵岳星
分析测试学报 2013年1期
张 勇,薛昆鹏,何 美,陈再洁,吴 虹,赵岳星
(1.安徽中医学院 药学院,安徽 合肥 230031;2.月旭材料科技(上海)有限公司,上海 201203)
龙胆泻肝丸是名方龙胆泻肝汤的一种制剂,由龙胆、栀子、黄芩、泽泻等10味药物组成,具有泻肝胆、利湿热等功效,主治肝胆实火上扰。药方中龙胆为君,栀子、黄芩为臣[1],有效成分分别为龙胆苦苷、栀子苷和黄芩苷(化学结构式如图1),其中龙胆苦苷和栀子苷均属于环烯醚萜苷类成分,其化学结构非常相近,用高效液相色谱(HPLC)进行检测时易相互干扰[2]。
目前2010版《中国药典》1部中的龙胆泻肝丸已收载了3个成分的含量测定方法[3],然而,由于本品成分复杂,测定时不仅易产生干扰和误差,其所含的非指标性成分在采用HPLC测定时保留时间较长(全程60 min),影响其3个成分的快速、准确测定。因而,本研究利用固相萃取(SPE)技术对本品进行预处理后采用超高效液相色谱(UPLC)法进行测定[4],使分析时间降低至13 min,且所得峰形对称,便于保护UPLC细粒径色谱柱。该方法简便易行,可更好、更快地用于该制剂的质量控制。
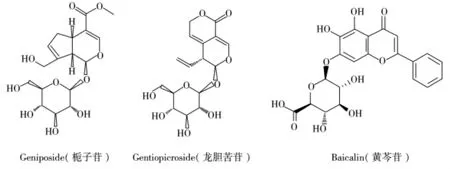
图1 栀子苷、龙胆苦苷和黄芩苷的化学结构式Fig.1 Chemical structures of geniposide,gentiopicroside and baicalin
1 实验部分
1.1 仪器与试剂
Acquity H-Class UPLC超高效液相色谱仪、Empower 2色谱数据工作站(美国Waters公司),Milli-Q超纯水制备系统(美国Millipore公司),CP225D型电子天平(德国Sartorius公司),KQ-200KDB超声波提取器(220 V,50 kHz),WelchromC18E固相萃取柱(200 mg/3 mL,月旭科技公司)。
龙胆泻肝丸(水丸,规格:6 g/100粒;批号:0083081、0083082、0083083,北京同仁堂制药有限公司);龙胆苦苷对照品(110770-201013)、栀子苷对照品(110749-200714)、黄芩苷对照品(110715-201016)购自中国药品生物制品检定所;甲醇、乙腈(色谱纯,美国天地公司)。
1.2 标准溶液的配制
精密称取栀子苷、龙胆苦苷和黄芩苷对照品各0.80、1.07、2.05 mg,置于25 mL容量瓶中,用甲醇溶解并稀释至刻度,摇匀,即得对照品混合溶液。
1.3 样品前处理
取适量龙胆泻肝丸,研细,分取两份,按照如下步骤制备样品溶液。
1.3.1 样品的提取 精密称定研好的粉末1 g,置于具塞锥形瓶中,精密加入50 mL 50%甲醇水溶液,盖紧瓶塞,称定重量,超声处理20 min,放冷,用50%甲醇补足重量,摇匀,过滤,弃去初滤液,取续滤液,得样品提取液,待净化。
1.3.2 样品的净化 依次用甲醇、超纯水和50%甲醇各5 mL活化C18固相萃取柱[5],随后精密移取5 mL上述处理好的样品提取液加载到活化后的C18柱上,控制流速为1滴/s,弃去初滤液2.5 mL,收集后续的流出液,经0.22 μm有机微孔滤膜过滤,以备测定。
1.4 仪器条件
2 结果与讨论
2.1 色谱分离体系的优化
采用UPLC对目标物质进行快速分析,选择1.8 μm粒径的C18色谱柱,发现当选择2.1 mm×50 mm和2.1 mm×100 mm两种型号色谱柱时,栀子苷和龙胆苦苷很难分离,而选择4.6 mm×50 mm色谱柱,样品可达到基线分离,原因可能是由于中药样品基质复杂,选择太细的色谱柱,填料量过少使得样品过载而无法分离;流动相参照2010版中国药典1部及其他文献进行对比分析[2-3,6-8],发现在甲醇-0.2%磷酸体系下,峰形拖尾严重;在乙腈-1%醋酸体系下,栀子苷和龙胆苦苷无法达到基线分离;而选用乙腈-0.1%磷酸体系时,所得峰形较好,分离度满足要求,且分析总时间相比于药典方法缩短了一半(见图2),并且药典方法选用的流动相(0.2%磷酸溶液)pH值过低(pH<2.0),不适于色谱柱的长期使用。
2.2 流动相条件的优化
以不同比例的乙腈-0.1%磷酸水溶液进行初始梯度设置,发现当乙腈初始含量低于12%时,龙胆苦苷和栀子苷的保留时间均会延后,且分离度随之下降,当乙腈达5%时两峰完全重叠。而乙腈初始含量高于12%时,两峰的分离度虽然有所提高,但出峰太快,导致与前面峰难以分离,因此,选择12%乙腈-0.1%磷酸水溶液作为最佳初始流动相。
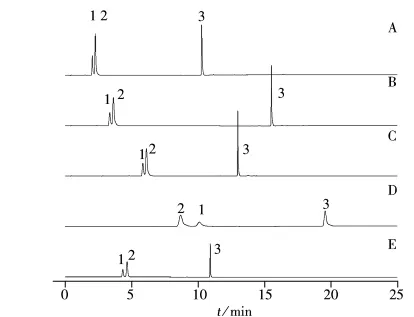
图2 3种化合物在不同分离体系下的色谱图Fig.2 Comparison of chromatograms of three compounds under different systems
2.3 样品前处理的优化
考察了直接提取和使用SPE净化两种前处理方式时目标物的分离效果。结果表明:直接提取的样品,需在11.5 min后渐进提高乙腈比例至50%,才能将黄芩苷出峰后的非指标性成分洗脱,而采用SPE净化后的样品,由于去除了大量杂质的干扰,不需要用高比例有机相进行后续杂质的洗脱,大大缩短了平行进样梯度系统平衡的时间。
2.4 SPE净化条件的优化
本实验将50%甲醇提取液直接经C18固相萃取柱进行净化,在此洗脱溶剂系统下,每隔0.5 mL收集流出液,共收集9管。以流出体积为横坐标,3个成分的吸收峰面积为纵坐标作流出曲线,结果在第6管即初滤液在2.5 mL后,流出液的栀子苷、龙胆苦苷和黄芩苷的峰面积已达平衡,因此最终选择流出液的初滤液体积为2.5 mL。这主要是利用待测成分在样品液与C18固定相之间达成分配平衡后,流出液与样品液中所含待测成分的浓度一致,而此时样品溶液中对C18分配系数较大的成分仍保留在萃取柱中,从而达到排除干扰物质、减少分离时间和保护色谱柱的目的[9]。为考察待测组分的饱和流出曲线受洗脱溶剂极性的影响,将样品提取液的甲醇含量分别调至40%和60%进行平行洗脱,发现在40%甲醇洗脱溶剂下,10 mL内无法达到3种待测组分的流出平衡,而在60%甲醇洗脱溶剂下,柱上强保留的杂质均洗脱下来,对黄芩苷的分离产生干扰。综合考虑,选择50%甲醇作为SPE的洗脱溶剂。
2.5 流速与柱温的优化
考察了不同柱温和流速对各组分分离度的影响,结果见表1。由表1可知:流速为0.7 mL/min时,栀子苷与龙胆苦苷的分离度较好,但黄芩苷与前后峰的分离度不理想;在相同条件下流速增加到0.8 mL/min时,3种主要成分的分离度均较佳,且保留时间缩短;当流速增加到0.9 mL/min时,三者的分离度未改善,反而有所降低。柱温为20℃时,栀子苷与龙胆苦苷能得到良好分离,黄芩苷与前一个杂质峰分离较好,与其后的杂质峰分离度基本满足定量要求;柱温升至25℃时,各主要成分的分离度均大于1.5,符合要求;当柱温为30℃时,各组分的分离度均降低。综合考虑,流速选择0.8 mL/min,柱温选择25℃。
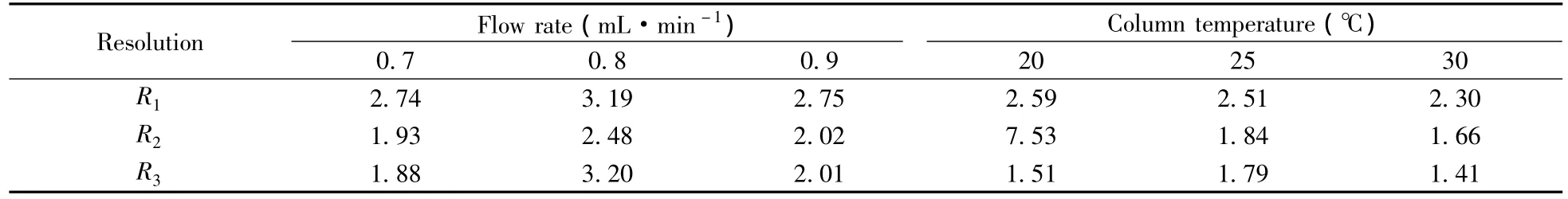
表1 3种成分在不同流速和不同柱温下的分离度Table 1 Resolutions of three compounds under different flow rates and column temperatures
2.6 方法的精密度、线性范围、检出限与定量下限
取“1.2”对照品混合标准溶液,按本方法进行分析,于同日内测定6次,连续测定5日,测得栀子苷、龙胆苦苷和黄芩苷峰面积的日内和日间精密度;将对照品混合溶液分别进样0.4、0.6、1.0、2.0、3.0、4.0 μL进行测定,以峰面积(Y)对标样的进样量(X,ng)进行线性回归,得到各化合物的线性回归方程;并以3倍信噪比(S/N=3)所对应的浓度为检出限(LOD),以10倍信噪比(S/N=10)对应的浓度为定量下限(LOQ),结果见表2。从表2可看出,栀子苷在12.8~128 ng,龙胆苦苷在17.2~172 ng和黄芩苷在32.8~328 ng范围内呈良好线性,相关系数均大于0.999,日内和日间相对标准偏差均不超过1.8%。
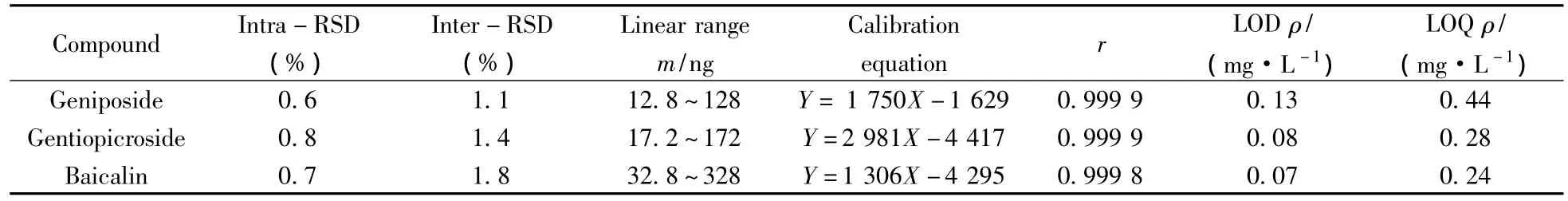
表2 方法的精密度、线性范围、检出限及定量下限(n=6)Table 2 Precision,calibration curve,LOD and LOQ of the method(n=6)
2.7 方法的重复性、稳定性及专属性
精密称取同一批号龙胆泻肝丸1 g,按“1.3.2”方法制备6份供试品溶液进行测定,得到栀子苷、龙胆苦苷和黄芩苷的RSD分别为1.7%、0.8%和0.7%。
重新制取一份供试品溶液,分别在0、1、2、4、8、12 h进行测定,3个成分的 RSD分别为0.7%、1.2%和0.9%,表明12 h内3个成分在供试品溶液中基本稳定。
按处方比例分别制取缺栀子、缺龙胆和缺黄芩的阴性样品,精密称取1 g,按照“1.3.2”方法,制备阴性样品溶液后进行测定,色谱对比图见图3。结果显示对栀子苷、龙胆苦苷和黄芩苷的含量测定无干扰,专属性良好。
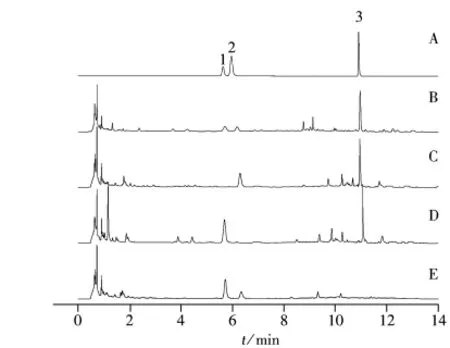
图3 混合对照品(A)、供试品(B)、缺栀子阴性样品(C)、缺龙胆阴性样品(D)、缺黄芩阴性样品(E)的UPLC色谱图Fig.3 UPLC chromatograms of reference substances(A),sample(B),negative sample without Gardeniae Fructus(C),negative sample without Gentianae Radix et Rhizoma(D),negative sample without Scutellariae Radix(E)1.geniposide,2.gentiopicroside,3.baicalin
2.8 回收率实验
精密称取龙胆泻肝丸(批号:0083081)粉末0.5 g 6份,分别精密加入对照品混合溶液(另配,栀子苷0.82 g/L,龙胆苦苷0.64 g/L,黄芩苷2.31 g/L)1 mL,按“1.3.2”方法制备溶液后测定,计算得栀子苷、龙胆苦苷和黄芩苷的平均回收率为99%、99%和95%,RSD分别为2.3%、1.9%和2.8%。黄芩苷回收率偏低可能是由于C18萃取柱填料对于分配系数大的组分部分吸附所致。
2.9 实际样品的测定
分别取不同批号的龙胆泻肝丸,按“1.3.2”方法处理,每批样品制备3份,用标准曲线法分别计算3批样品各成分的含量,测定结果见表3,该结果符合中国药典含量测定规定。为进一步进行方法学验证,采用现行国家药品标准的常规HPLC方法,选择岛津LC-10A液相色谱仪,Ultimate AQ-C18(4.6 mm×250 mm,5 μm)柱,甲醇-0.2%磷酸梯度洗脱对3批龙胆泻肝丸样品进行检测,每个样品平行测定3次,测定结果见表3。数据显示,药典法测定结果与本法测定结果具有良好的一致性。对两种方法的检测结果进行F值和t值检验,结果显示,两种方法之间无显著性差异。此外,药典法所需分析时间为60 min,流动相的pH值小于2.0,对栀子苷、龙胆苦苷和黄芩苷的定量下限分别为6.84、8.79、4.24 mg·L-1,相比于该方法,本实验所建立的方法分析时间更短,灵敏度更高。
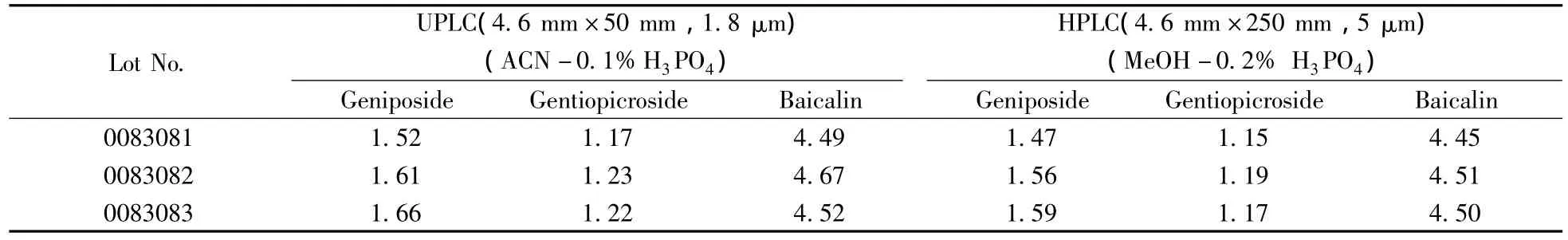
表3 两种方法测定龙胆泻肝丸含量数据对比Table 3 Determination results of different experimental methods in Longdanxiegan Pill w/(mg·g-1)
3 结论
本实验建立的SPE/UPLC方法,可在较短时间内实现对龙胆泻肝丸中栀子苷、龙胆苦苷和黄芩苷三个指标成分的快速检测。该方法灵敏度高、重复性好,可为今后该制剂的质量控制提供理论依据。
[1]Xu J,Peng H,Yang W L.Chin.Tradit.Herbal Drugs(许军,彭红,杨武亮.中草药),1999,30(5):378-379.
[2]Hao F,Jiang Y,Li Y R,Ding X Y,Hu L Y.Chin.Tradit.Herbal Drugs(郝福,蒋晔,李艳荣,丁翔宇,胡丽英.中草药),2006,37(12):1808-1809.
[3]The Pharmacopoeia of the People's Republic of China.Vol 1.State Pharmacopoeia Committee of China(中国药典1部.国家药典委员会),2010:636-637.
[4]Meng J,Wang S F,Han F,Li S M.Chin.J.Chin.Mater.Med.(孟健,王淑芬,韩飞,李三鸣.中国中药杂志),2006,31(2):119-121.
[5]Fang Z E,Qu F,Shang J C.J.Instrum.Anal.(方志娥,曲斐,尚京川.分析测试学报),2010,29(3):276-279.
[6]Tian S X,Jiang Y,Hao F.Chin.Tradit.Patent Med.(田书霞,蒋晔,郝福.中成药),2006,28(11):1578-1581.
[7]Liu R X.Chin.Tradit.Patent Med.(刘瑞新.中成药),2007,29(8):1170-1172.
[8]Liu H,Su J,Liang X,Zhang X,He Y J,Huang H Q,Ye J,Zhang W D.J.Pharm.Anal.,2011,1(1):1 -7.
[9]Chen Y,Zhu B H,Fang J H.Chin.Tradit.Patent Med.(陈勇,朱炳辉,方继辉.中成药),2002,24(10):761-764.
猜你喜欢
基层中医药(2022年7期)2022-11-17 08:25:02
现代畜牧科技(2021年8期)2021-10-13 07:21:56
今日农业(2020年16期)2020-12-14 15:04:59
基层中医药(2020年7期)2020-09-11 06:38:06
基层中医药(2018年7期)2018-12-06 09:25:50
中国现代中药(2018年11期)2018-11-19 05:24:08
中成药(2017年12期)2018-01-19 02:06:54
中成药(2017年12期)2018-01-19 02:06:43
中成药(2016年4期)2016-05-17 06:07:40
金色年华(2016年11期)2016-02-28 01:42:20
