
人耳聋相关基因GJB3的结构特征及生物信息学分析
2013-10-29 09:36:54孙丹王帅魏钦俊姚俊曹新
生物技术通讯 2013年1期
孙丹,王帅,魏钦俊,姚俊,曹新
南京医科大学 基础医学院,江苏 南京 210029
听力障碍是人类最常见的感觉神经紊乱,世界范围约1000个儿童就有1例患有听力障碍。在中国,每年2000万的新生儿中有30 000例患有先天性耳聋[1]。已经明确,在引起耳聋的各类原因中,遗传因素致聋已逐渐成为主导因素,在工业化国家至少50%耳聋病例都是遗传因素所致[2]。遗传性耳聋分为综合征型耳聋(syndromic hearing loss,SHL)和非综合征型耳聋(non-syndromic hearing loss,NSHL),其中NSHL在耳聋儿童中的发生率为80%[3-5]。NSHL具有全部的孟德尔遗传方式及线粒体基因的母系遗传方式,其中常染色体隐性遗传方式约占77%,常染色体显性遗传方式约占22%,X连锁和线粒体遗传占1%~2%[6-7]。迄今,已明确超过60种基因与感音神经性耳聋有关,还存在上百个听力损伤相关的基因座[8]。
间隙连接蛋白(connexin,Cx)基因是一类最常见的与遗传性耳聋相关的致聋基因。Cx是间隙连接通道的亚单位蛋白,它允许离子和相对分子质量小于1500的代谢物在细胞间扩散。介导质膜间通讯的2个半通道又称为连接子,它的作用是彼此对接形成间隙连接通道,从而实现间隙连接细胞间的通 讯(gap functionalintercellularcommunication,GJIC)[9]。GJIC在维持细胞内环境稳定中具有十分重要的作用,可通过相邻细胞的相互调控及物质交换促进细胞正常生长与分化,当其功能下调或丧失时,导致包括耳聋在内的多种疾病的发生[10]。
在耳蜗毛细胞,GJIC主要涉及3种Cx,即Cx26、Cx31和Cx30。编码Cx26的GJB2基因是遗传性NSHL最常见的耳聋基因,其突变类型广泛且突变率高[11-12]。而编码Cx30、Cx31的GJB6、GJB3与GJB2相比,无论在突变谱还是致病性突变频率上,均表现出明显的差异性。因此,深入研究这2类基因的分子结构与进化特征,对了解该基因家族突变致聋的分子机制是十分有益的。
我们利用生物信息学方法,对人类GJB3基因的编码区序列结构进行了分析和预测,为该基因后续相关功能和致病性研究奠定了基础。
1 材料与方法
1.1 材料
选择17个物种的Cx31蛋白质序列(表1),长度为257~304残基。这17个物种分别来自哺乳纲、鸟纲、两栖纲和鱼纲,其中9个物种的Cx31蛋白质序列为预测序列。
生物信息分析软件及在线分析工具包括多序列比对软件ClustalW2(http://www.ebi.ac.uk/Tools/msa/clustalw2/)、进化分析和进化树绘制软件MEGA 5.05(http://www.megasoftware.net/)、蛋白质三级结构预测工具SWISS-MODEL(http://swissmodel.expasy.org/)、蛋白质模建构图软件 VMD1.9(http://www.ks.uiuc.edu/Research/vmd/vmd-1.9/)。
1.2 GJB3进化树绘制与分析
将17个物种(表1)的Cx31序列用MEGA5.05进行多重序列比对,并选择与Cx31同为缝隙连接蛋白家族的人类Cx30作为外群(outgroup)建立NJ(Neighbor joining)树,用Test of Phylogeny(发育检验方法)选择 Bootstrap method,Bootstrap Replica⁃tions(Bootstrap检验次数)选择1000,采用泊松模式建树以获得系统发育结论。
1.3 GJB3基因保守性分析
选取Cx31系统发育树中哺乳动物纲除鸭嘴兽外各末端节点处物种1种(本研究选择了人类、荷兰猪、家犬、家鼠、中国仓鼠等5个物种),用ClustalW2软件对GJB3基因编码蛋白进行多序列比对,计算保守位点百分数。
1.4 Cx31蛋白三维结构分析
将Cx31蛋白质氨基酸序列提交至SWISSMODEL进行模建,得到的pdb文件用VMD1.9软件进行可视化分析。依据SWISS-MODEL提供的自动化比较蛋白质模建服务器和SWISS-MODEL库进行同源模建[13-15]。
1.5 GJB3基因错义突变分析
为研究GJB3发生错义突变后三维结构的改变,选择了2种错义突变(p.E183K和p.I141V)进行三维结构突变前后的比较,并分析了Anolea原子平均势能。采用同源模建软件SWISS-MODEL模建Cx31蛋白的三维结构,用VMD1.9软件对突变前后的三维结构进行可视化分析,最后通过Anolea原子平均势能图的改变评价错义突变对Cx31蛋白三维结构的影响。
2 结果
2.1 GJB3系统发育分析
用MEGA5.05构建的Cx31的NJ树见图1。进化树中哺乳纲、鸟纲、两栖纲和鱼纲动物分界明晰,符合物种进化从水生到陆生的过程。其中哺乳纲动物除鸭嘴兽外其余各物种亲缘关系接近,这与鸭嘴兽为最原始的哺乳动物有关;另从图中可见,两栖动物与鱼类的亲缘关系更为接近,这验证了鱼类为两栖类动物的祖先这一点。该NJ树的构建以与Cx31同家族的人类Cx30蛋白为外群,Blast两两比对显示Cx31和Cx30的同源相似度仅为45%,且该树中Cx30成独立一支,说明选择Cx30作为外群是合理的。外群的构建增加了系统进化树的置信度。

表1 各物种的Cx31蛋白质序列信息
2.2 GJB3保守性分析

图1 Cx31蛋白的系统发育树

图2 Cx31多序列比对结果
如图2,用ClustalW2对GJB3编码的蛋白质Cx31进行多序列比对,结果显示Cx31完全保守和高度保守残基的位点占全部氨基酸残基的83.70%(226/270)。将Cx31的序列提交至PredictProtein后可得到氨基酸位点对应的各区域预测。预测结果显示Cx31蛋白有4个跨膜区(M1、M2、M3、M4)、2个胞外环(E1、E2)和3个胞浆区(N-term、CL、C-term)。经数据分析,跨膜区平均保守度为92.31%(72/78);胞外环平均保守度为94.12%(64/68);胞浆区平均保守度为72.00%(90/125),其中胞浆区C端保守度极低,仅为53.97%(34/63)(表2)。
2.3 Cx31三维结构分析
同源建模后的pdb坐标文件用VMD1.9对Cx31蛋白进行可视化,分析可知Cx31有6个α螺旋、3个β折叠、7个转角、7个无规则卷曲。Cx31蛋白质三维结构如图3所示,M1~M4分别为4个跨膜区。由于Cx31的同源建模是以Cx26为模板的,Cx26氨基酸残基数目少于Cx31,所以即便Cx31存在270个氨基酸残基,SWISS-MODEL仅提供2~212位氨基酸残基的蛋白质结构预测模型。也就是说,大部分的胞浆区C端没有被预测出来,但其他区域的结构都得到了很好的预测。
2.4 GJB3错义突变分析
GJB3致病性错义突变很少,耳聋变异数据库[Deafness Variation Database(http://deafnessvaria⁃tiondatabase.com/)]共收录了9种导致NSHL的GJB3错义突变(表3)。
我们选择Cx31位于跨膜区M1的p.Arg32Trp突变和位于胞外环E2的p.Glu183Lys突变,对Cx31跨膜区和胞外环突变导致NSHL的发生机制分别进行分析。M1区32位Arg被Trp替代,Anolea原子平均势能图显示了4个明显变化区域(图4),各原子势能的增加可导致CX31结构由低能量的稳态转变为较高能量的不稳态。用VMD1.9对CX31进行突变前后的结构可视化,发现Anolea平均势能图4个明显变化区域分别为跨膜区M1、M2、M3、M4,且4个变化区域的氨基酸残基均与M1区32位氨基酸残基空间结构上接近(局部三维结构见图4A,a和b)。对Cx31分子结构进行重叠分析(图4A,c),发现193~194位 Gly和 Ala,196~198位 Ala、Val和 Cys空间结构位置发生了较明显的改变(蓝色为突变前残基位置,红色为突变后残基位置)。由此推测,位于Cx31跨膜区的某氨基酸残基改变可导致该蛋白其他跨膜区相近残基空间结构的改变,最终导致Cx31无法发挥其正常功能。

表2 Cx31结构域保守度分析

表3 已报道的NSHL的GJB3错义突变
E2区183位Glu被Lys替代,Anolea原子平均势能图上可观测到2个明显改变区域(图5),即E1区39~43位及E2区182~183位。在VMD1.9可视化中可看出这2个区域在空间结构上非常接近,即可推测183位Glu被Lys替代后影响了E1和E2区局部分子的分子间作用力。Xia等对Cx31错义突变的实验研究表明p.Glu183Lys会导致Cx31蛋白大部分滞留在高尔基复合体,少部分在内质网中,而在细胞与细胞间连接上没有发现[16]。因此,可推测发生在E2区的错义突变会导致Cx31不能从高尔基复合体运输到细胞膜上参与构建细胞间的间隙连接。
3 讨论
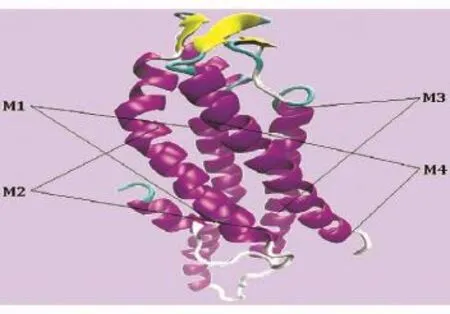
图3 Cx31三维结构预测结果

图4 野生型和p.Arg32Trp型Cx31蛋白的三维结构和Anolea平均势能比较
间隙连接蛋白可在相邻细胞间形成间隙连接通道,从而介导小分子物质的通讯。间隙连接蛋白在耳蜗Corti氏器的K+循环通路中发挥着至关重要的作用。K+流入毛细胞机械门控离子通道后,毛细胞将去极化,接着Ca2+流入基底膜,介导神经递质释放。间隙连接蛋白的丢失将会导致K+循环被破坏,Corti氏器局部K+浓度过高发生钾中毒。GJB3基因编码的蛋白Cx31是间隙连接蛋白家族中的重要成员,分布在身体多个组织器官上,但以皮肤和内耳最多,其突变可导致可变性红斑皮肤角化病(EKV)、听力损伤和周围神经病变[17-18]。

图5 野生型和p.Glu183Lys型Cx31蛋白三维结构和Anolea平均势能比较
为了更好地理解GJB3基因突变与NSHL发生的相关性,我们利用生物信息学软件,从GJB3系统发育、保守性、三维结构和错义突变角度分别进行了预测分析。Cx31在跨膜区和胞外环的保守性远高于胞浆区,胞浆区C端保守度极低。Cx26和Cx31均为间隙连接蛋白家族成员,但GJB3基因致病性突变远少于GJB2基因。对GJB2编码蛋白Cx26的保守性分析显示其保守度为95.58%(216/226),而同物种多序列比对显示Cx31保守度为83.70%(226/270)。因此,Cx31的保守性低于Cx26,突变对Cx31的影响要小于Cx26,可推测这是GJB2突变致聋的人群远多于GJB3突变原因之一。对已报道的p.Arg32Trp和p.Glu183Lys两类错义突变,对其Cx31跨膜区和胞外环氨基酸发生改变后三维结构改变和Anolea原子平均势能变化的分析,将推进从Cx31蛋白分子结构上对GJB3错义突变致病的分子机制的理解。我们以2种突变为例,发生在跨膜区的p.Arg32Trp可能会导致其他几个跨膜区氨基酸空间结构的改变而使得Cx31无法发挥其正常功能;同时,结合Xia等的实验研究,发生在胞外环的p.Glu183Lys使得Cx31蛋白不能完成高尔基复合体、内质网上的加工修饰而正常定位于细胞膜上,无法介导正常的细胞间连接通讯。由这2种突变可推测,GJB3基因发生错义突变后,其编码的Cx31结构将会发生改变,难以定位于细胞膜上,不能发挥间隙连接的功能。
虽然通过生物信息学方法对基因和蛋白质进行分子结构与功能的预测分析依旧有其局限性,但这些工作对于生物分子系统的生物学研究起到了很好的前瞻性作用。我们利用生物信息学方法首次对GJB3基因的编码区进行了系统发育树构建、保守区多序列比对分析及三维结构与某些错义突变功能分析,为今后相关的实验研究提供了有价值的信息并奠定了重要的基础。
[1]Yuan Y,You Y,Huang D,et al.Comprehensive molecular etiology analysis of nonsyndromic hearing impairment from typ⁃ical areas in China[J].J Transl Med,2009,7:79-90.
[2]Rodriguez-Paris L,Pique L,Colen T,et al.Genotyping with a 198 mutation arrayed primer extension array for hereditary hearing loss assessment of its diagnostic value for medical practice[J].PLoS One,2010,5(7):e11804.
[3]Adato A,Raskin L,Petit C,et al.Deafness heterogeneity in a Druze isolate from the Middle East:novel OTOF and PDS mutations,low prevalence of GJB2 35delG mutation and indi⁃cation for a new DFNB locus[J].Eur J Hum Genet,2000,8(6):437-442.
[4]Varga R,Avenarius M R,Kelley P M,et al.OTOF muta⁃tions revealed by genetic analysis of hearing loss families in⁃cluding a potential temperature sensitive auditory neuropathy allele[J].J Med Genet,2006,43(7):576-581.
[5]Kim S,Song D G,Bae J W,et al.A family of H723R muta⁃tion for SLC26A4 associated with enlarged vestibular aque⁃duct syndrome[J]. Clin Exp Otorhinolaryngol, 2009,2(2):100-102.
[6]Yasunaga S,Grati M,Chardenoux S,et al.OTOF encodes multiple long and short isoforms:genetic evidence that the long ones underlie recessive deafness DFNB9[J].Am J Hum Genet,2000,67(3):591-600.
[7]Adato A,Raskin L,Petit C,et al.Deafness heterogeneity in a Druze isolate from the Middle East:novel OTOF and PDS mutations,low prevalence of GJB2 35delG mutation and indi⁃cation for a new DFNB locus[J].Eur J Hum Genet,2000,8(6):437-442.
[8]Frenzel H,Bohlender J,Wohlleben K,et al.A genetic basis for mechanosensory traits in humans[J].PLoS Biol,2012,10(5):e1001318.
[9]Schnichels M,Wörsdörfer P,Dobrowolski R,et al.The con⁃nexin31 F137L mutant mouse as a model for the human skin disease erythrokeratodermia variabilis(EKV)[J].Hum Mol Gen⁃et,2007,16(10):1216-1224.
[10]Lee H J,Rhee S K.Growth-suppressing activity of the trans⁃fected Cx26 on BICR-M1Rk breast cancer cell line[J].J Mi⁃crobiol Biotechnol,2011,21(5):477-482.
[11]Liu X Z,Yuan Y,Yan D,et al.Digenic inheritance of non-syndromic deafness caused by mutations at the gap junc⁃tion proteinsCx26 and Cx31[J].Hum Genet,2009,125(1):53-62.
[12]Gardner P,Oitmaa E,Messner A,et al.Simultaneous multi⁃gene mutation detection in patients with sensorineural hearing loss through a novel diagnostic microarray:a new approach fornewborn screening follow-up[J].Pediatrics,2006,118(3):985-994.
[13]Arnold K,Bordoli L,Kopp J,and et al.The SWISS-MODEL Workspace:a web-based environment for protein structure ho⁃mology modelling[J].Bioinformatics,2006,22:195-201.
[14]Schwede T,Kopp J,Guex N,et al.SWISS-MODEL:an auto⁃mated protein homology-modeling server[J].Nucleic Acids Res,2003,31:3381-3385.
[15]Guex N,Peitsch M C.SWISS-MODEL and the Swiss-Pdb⁃Viewer:an environment for comparative protein modelling[J].Electrophoresis,1997,18:2714-2723.
[16]Xia K,Ma H,Xiong H,et al.Trafficking abnormality and ER stressunderlie functional deficiency of hearing impair⁃ment associated connexin-31 mutants[J].Protein Cell,2010,1(10):935-943.
[17]Minárik G,Ferák V,Feráková E,et al.High frequency of GJB2 mutation W24X among Slovak Romany(Gypsy)patients with non-syndromic hearing loss(NSHL)[J].Gen Physiol Bio⁃phys,2003,22:549-556.
[18]Wang W H,Liu Y F,Su C C,et al.A novel missense muta⁃tion in the Connexin30 causes nonsyndromic hearing loss[J].PLoS One,2011,6(6):e21473.
猜你喜欢
广东药科大学学报(2023年5期)2023-12-30 00:08:39
中学生数理化·八年级物理人教版(2023年6期)2023-05-25 11:59:48
——《势能》
文化纵横(2022年3期)2022-09-07 11:43:18
生物化学与生物物理进展(2022年6期)2022-07-21 11:52:06
中学生数理化·八年级物理人教版(2022年6期)2022-06-05 06:55:40
时代报告·奔流(2022年1期)2022-04-29 04:10:56
昆明医科大学学报(2022年3期)2022-04-19 13:59:42
中学生数理化·八年级物理人教版(2021年6期)2021-11-22 07:49:52
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19 08:52:38
池州学院学报(2015年3期)2016-01-05 01:13:04
