
甲基环己烷催化裂化的研究进展
2013-10-24 12:30郭锦标王鑫磊
石油化工 2013年1期
张 旭,周 祥,郭锦标,王鑫磊
(中国石化 石油化工科学研究院,北京 100083)
近年来世界原油不断向重质化和劣质化发展,给石油炼制工业带来了许多新的问题。沥青合成原油中芳烃和环烷烃的含量远高于常规原油,达到70%(w)[1]。加工高芳烃和环烷烃含量的原油,将是各国炼油工业共同面对的挑战和发展趋势。众所周知,高芳烃含量的原油不但会降低催化裂化过程的转化率,减少汽油、柴油和其他中间馏分的产量,而且催化剂容易结焦并最终降低催化裂化装置的性能[2]。环烷烃易发生脱氢和氢转移反应生成芳烃[3],是芳烃和焦炭的前体[4]。环烷烃由于其特殊的化学结构,在催化裂化反应中显示出了不同于烷烃和芳烃的反应行为。
目前对单体烃的催化裂化反应研究较多,如链烷烃[5]、环烷烃[6]和芳烃[7]等。通过模型化合物的催化裂化反应可以清楚地认识裂化过程中所发生的化学反应。由于甲基环己烷(MCH)与多环环烷烃具有一定的相似性[1],认识MCH的催化裂化转化规律,对进一步认识多环环烷烃和多环芳烃在催化裂化反应过程中的转化规律具有重要的参考意义,有助于认识催化裂化反应过程中的结焦现象,为工艺优化和催化剂研制提供理论支持。
本文主要对MCH催化裂化反应的催化剂、转化机理和动力学研究进行了综述;讨论了不同种类催化剂和工艺条件对MCH转化的影响,梳理了转化过程中的主要反应类型;在此基础上进一步总结了MCH在典型催化剂上的反应网络。
1 催化剂性质的影响
催化剂的孔道结构、比表面积、酸类型、酸浓度及其分布等对MCH的转化有很大的影响。表1列出了近年来国内外在MCH催化裂化方面的研究结果[8-19]。由表1可看出,研究重点集中在催化剂的结构性能、转化机理、动力学及工艺条件优化等方面。另外,表1还列出了甲基环戊烷[20-21]和环己烷[22-23]的催化裂化研究情况,研究的核心仍然是催化剂的构效关系和反应机理两个方面。

表1 单环环烷烃催化裂化研究现状Table 1 Research on the catalytic cracking of monocyclic cycloalkanes
前人对低相对分子质量环烷烃模型化合物的转化情况,如环己烷、甲基环戊烷和MCH等在不同催化剂上的催化裂化反应开展了大量的研究工作,获得了对环烷烃催化裂化反应的一些新认识。
Corma等[8]系统研究了500 ℃、常压条件下,MCH在REY和USY分子筛催化剂上的裂化情况。研究结果表明,可以结合β-断裂、质子裂化、异构化、氢转移和脱氢5种反应类型来解释MCH开环裂化反应,甲基环己烯是脱氢反应过程中的重要中间物种。Corma等[9]还考察了IM-5和ZSM-5分子筛催化剂在MCH催化裂化反应条件下的热稳定性和水热稳定性,IM-5分子筛比ZSM-5分子筛显示出更高的稳定性,可以获得更高的转化率和烯烃/烷烃比。
Cerqueira等[14]对比了MCH在不同硅铝比的HFAU,HBEA,HMFI沸石上的转化情况。C2~7烯烃、C4~7异构烷烃及甲苯是MCH的主要转化产物,在上述3种催化剂上,产物分布完全不同,在HMFI沸石上生成H2、CH4和苯,但在HFAU沸石上不生成。通过HFAU沸石上的产物分布可看出,MCH的转化遵循典型的碳正离子机理;而在HMFI沸石上,氢质子辅助脱氢起到了重要的作用。两者反应机理的不同与HMFI沸石上的狭窄孔道(即空间位阻)影响了氢转移有关。结合反应产物分布及反应过程中结焦的影响,得出了MCH的主要转化路径(见图1)[15,18]。
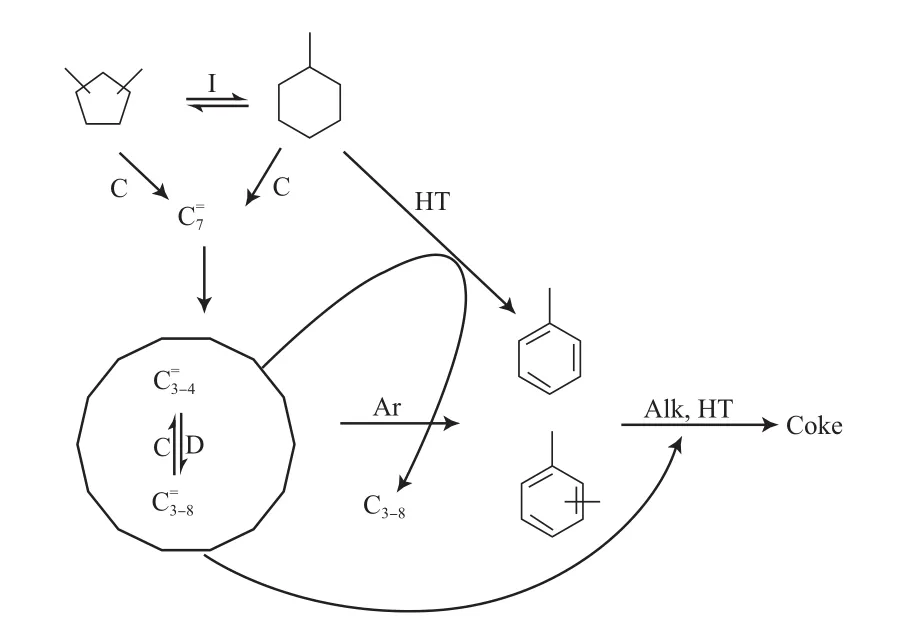
图1 MCH的主要反应路径Fig.1 Main reaction paths of MCH.
为进一步认识MCH在分子筛催化剂上的转化路径,前人对MCH分子在不同分子筛中的吸附、扩散和反应过程进行了详细研究。袁帅等[24-25]采用分子模拟的方法计算了不同类型石油烃分子的扩散能垒关系,单环的无取代基和短支链环烷烃可以在MFI和FAU分子筛孔道内进行扩散。当分子筛的孔道尺寸大于反应物分子尺寸时,分子可以自由扩散进入;当两者的尺寸相当时,反应物分子也可以进入孔道,但存在扩散阻力;当分子筛的孔道尺寸小于反应物分子尺寸时,分子无法进入孔道,只能在分子筛的外表面发生反应。图2是MCH稳定构型下的侧视图和俯视图,图中标明了MCH分子的长、宽、高数值[26]。
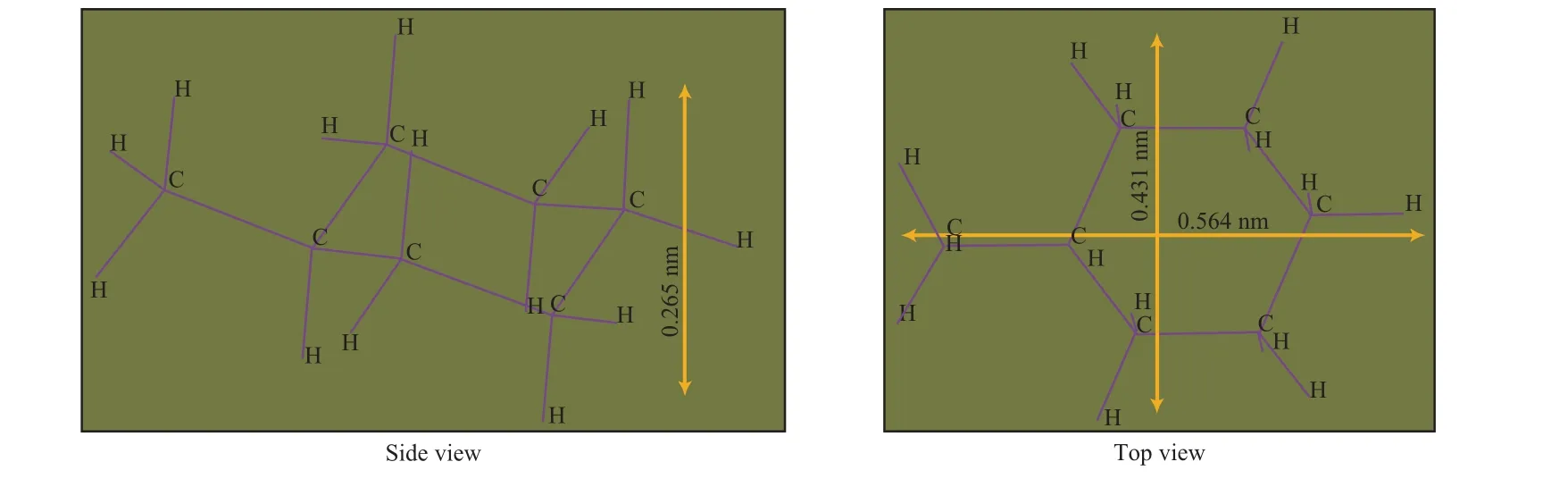
图2 MCH最低能量下的分子尺寸Fig.2 Two views of the minimum energy conformation of MCH.
Al-Sabawi等[17]发现晶粒大小对MCH在晶粒内的扩散没有影响。大晶粒分子筛由于有较高的酸度可以获得较高的MCH转化率,这与张领辉等[27]得到的关于不同晶粒大小的ZSM-5分子筛催化剂的裂化反应差异结论相吻合。0.9 μm的大晶粒分子筛有利于双分子氢转移和烷基转移反应,主要是因为大晶粒分子筛的孔道较长,反应物分子要扩散较长的距离才能到达反应中心。
对于FAU和MFI这一类分子筛,MCH裂化转化率与分子筛的酸性有关,而产物收率与催化剂的结构有关。即对某种特定的分子筛结构,MCH的裂化速率由酸性控制,而产物的选择性则由催化剂的结构决定[28]。
关于催化剂中各种孔道和活性位是如何影响MCH的转化路径的,也有相关文献报道[29]。HMCM-22 分子筛拥有2个互不相通的多维孔道体系。一条为二维正弦网状孔道,为十元环孔径;另一条同为十元环,但含有大的超笼。超笼沿水平剖开为十二元环,笼大小为0.71 nm×0.71 nm×1.82 nm。HMCM-22分子筛中各种孔道结构在MCH转化过程中所起的作用不尽相同[18],超过85%的MCH在超笼中发生反应,约11%的MCH在外层袋状孔穴中发生反应,只有不到2%的MCH在正弦孔道中发生反应,相应的转化频数(TOF值)分别为33,82,2 h-1。在催化剂各个位置反应生成的产物的分布有很大不同。可以运用碳正离子机理来解释超笼和袋状孔穴中形成的产物,而在狭窄的正弦孔道内需要质子化机理解释。在超笼中,反应活性高,结焦失活非常快;而其他位置的结焦失活可以忽略不计。
Scofield等[12]通过测量烯烃度(即产物中的烯烃量)来决定催化剂的空间位阻效应对氢转移反应的影响。对比不同结构的分子筛上MCH转化为烯烃的结果可看出,具有较为封闭孔结构的分子筛可以提供更高的烯烃选择性,而具有较大孔隙尺寸的沸石显示出较低的烯烃度,说明氢转移反应容易在较大孔径的分子筛上发生。在脱铝处理后的HBEA分子筛上MCH进行催化裂化反应时,脱铝处理对甲烷和甲苯的生成具有相同的效果,这意味着质子化作用对甲苯的生成具有重要的作用[30]。另外,MCH开环裂化形成大量的链烷烃,导致裂化产物的研究法辛烷值(RON)和马达法辛烷值(MON)较低;而MCH开环异构成五员环分子提高了裂化产物的辛烷值。因此,Santikunaporn等[11]提出通过适当的收缩环与开环催化剂的组合,即通过调变贵金属铂和HY沸石的含量和结构,可以最大限度地提高RON和MON。
2 工艺条件的影响
工艺条件(如反应温度、反应压力、剂油比和空速等)对MCH催化裂化反应也有一定的影响。研究发现,氢分压对MCH在酸性HZSM-5沸石上催化裂化的产物分布有一定的影响[13]。高氢分压增加了异构烷烃的含量,而4~6 MPa的氢分压提供了最佳的正构烷烃(乙烷、丙烷和正丁烷)选择性和低含量的甲烷(含量小于4%(w)),这正是蒸汽裂解的理想原料。不同的空速对转化率[19]和结焦[31]有很大影响,催化剂上焦炭的生成和分解与催化剂的孔结构和酸度有关,随接触时间的延长,结焦量也随之增加。
虽然有大量的单环和双环环烷烃在典型催化裂化催化剂上的转化研究报道,但大部分研究所选择的模型化合物的相对分子质量都小于实际的馏分油,并且鲜有对三环及三环以上的环烷烃和环烷芳烃纯组分的催化裂化反应报道;且文献报道也只是对环烷烃的宏观转化规律做定性描述,很少对其反应规律做出准确的描述。关于更高相对分子质量的环烷烃和环烷芳烃催化裂化[32-37]及开环反应[38-41]等的研究也有文献报道,在此不做详细介绍。
3 反应机理
MCH在沸石上的催化裂化反应主要是由氢转移、异构化、质子化开环裂化、β-断裂、脱烷基和烷基转移等6个主要反应所组成的复杂的串联、平行反应。
MCH分子包含5个仲碳原子、1个伯碳原子和1个叔碳原子,可以在环上发生开环反应,生成烷烃和烯烃,以H+进攻叔碳原子形成叔碳正离子为主,进一步发生β-断裂和开环反应等。在酸性催化剂上,环烷烃的六员碳环经过质子化环丙烷异构化,发生缩环反应,生成带有一个烷基取代的五员环环烷烃。另外,六员环具有较强的脱氢转化趋势,形成苯和甲苯等芳烃分子,芳烃进一步脱氢会形成焦炭。下面举例说明部分反应类型。
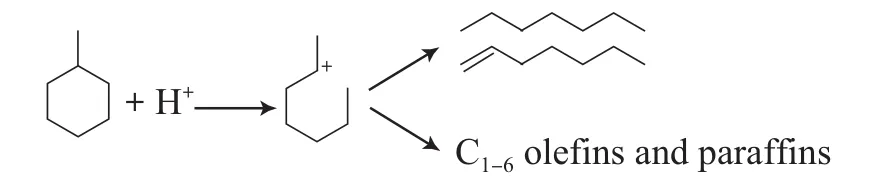
1)质子化开环反应
H+进攻活化MCH分子,经过一个五配位的碳正离子中间体,脱去一分子的氢气,形成稳定的叔碳正离子。甲基环己基叔碳正离子经过开环、β-断裂生成链烷烃和烯烃等。
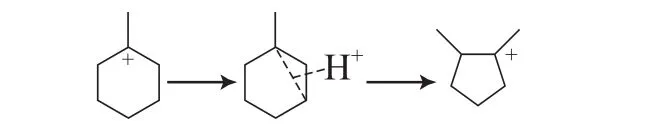
2)异构化反应
MCH可以异构为五员环的分子,如二甲基戊烷和乙基戊烷。由于环烷烃的环大小不同,其环张力大小也就不同。其中,五员环烷烃的环张力大于六员环[42],在催化裂化反应过程中具有较高的反应活性,很容易继续发生反应。六员环向五员环转变的异构化过程是MCH转化的一个重要过程,在相关文献中有广泛的报道[14,18]。
3)脱烷基
在MCH催化裂化反应中可以检测到少量的甲烷和环己烯分子,环己烯分子的存在可以由脱烷基反应来解释,Rabeharitsara等[19]对MCH脱烷基生成甲烷和环己烯的机理做了详细的说明。
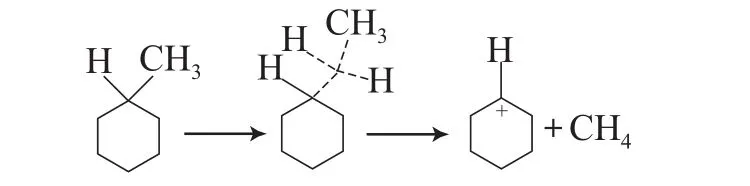
4)氢转移反应
MCH可以经过双分子氢转移反应形成甲苯等芳烃物质。在产物中有大量的芳烃生成,这也意味着在环烷烃反应过程中,双分子的氢转移反应是一个重要的反应步骤。事实上,在反应产物中,苯、甲苯和二甲苯等芳烃的含量约占30%(w)左右,远大于生成其他分子的选择性。

5)烷基转移
MCH经过两分子之间的烷基转移反应,生成C8和C6环烷烃,C8环烷烃有二甲基环己烷和乙基环己烷两种。另外,两分子甲苯会发生烷基转移反应,生成二甲苯和苯分子。

另外,MCH裂化生成的烯烃之间还会发生部分叠合反应,芳烃的深度脱氢会生成焦炭。
根据前人的研究结果和对MCH催化裂化反应的认识,总结了如图3所示的反应网络。

图3 简化的MCH催化裂化反应网络Fig.3 Simplified reaction network for MCH catalytic cracking.
MCH在转化的过程中主要涉及环烷烃的开环、β-断裂、异构化、烷基转移、脱烷基以及氢转移等六大类反应类型。具体来说,MCH受到催化剂酸性中心的活化,首先生成环烷烃上的碳正离子,进一步发生β-断裂和异构化反应,形成链状的烯烃、烷烃及部分五员环分子;MCH可以发生连续脱氢反应,形成甲苯,随氢转移反应深度的进一步加深,还可以生成焦炭;MCH分子之间还可以发生甲基转移,在氢转移的作用下,形成二甲苯和苯;MCH在质子氢的作用下还可以脱去取代基形成甲烷,环己基碳正离子则经过多步的连续脱氢,最终形成苯分子。
4 反应动力学
MCH催化裂化反应非常复杂,实验中检测到的产物多达40种以上。为了更好地为催化裂化的工艺设计优化提供基础数据及认识多环环烷烃和多环环烷芳烃的动力学转化规律,很多研究者对MCH的催化裂化动力学进行了研究。
建立MCH转化动力学模型是一个十分复杂的过程。因此,采取了集总方法来建立MCH转化的动力学模型。Al-Sabawi等[16]根据实验结果,综合考虑了吸附扩散过程和结焦的影响,根据关键组分,建立了16参数的动力学模型,通过非线性回归的方法求取动力学参数。图4所示的是基于集总水平的MCH转化过程动力学模型。
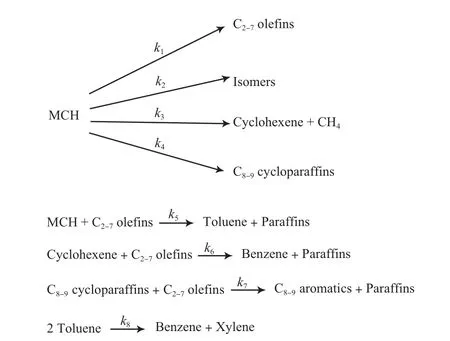
图4 MCH催化裂化过程的动力学模型Fig.4 Proposed kinetic model for MCH catalytic conversion.
对比活化能(Ea)可看出:Ea(3)>Ea(6)>Ea(1)>Ea(2)>Ea(4)>Ea(7)>Ea(5)>Ea(8)。低温有利于氢转移反应;而高温有利于开环反应。在MCH的转化过程中,相对于烷基转移和脱烷基反应,开环裂化、氢转移和异构化是主要反应类型。这与实验中得到的主要组成:C2~7烯烃、芳烃和MCH的五员环异构体结果相一致。由于MCH分子尺寸较小,容易进入分子筛孔道,因此,扩散过程对于MCH进入分子筛、到达活性中心参与反应没有影响。MCH在USY分子筛上的吸附是一个放热过程,吸附热达到了-40 kJ/mol。
Singh等[43]根据MCH的高温催化裂解反应,建立了一个完整的机理模型,编写了122个反应方程式,其中包含4个链引发、38个氢转移、6个甲基转移、11个叠合反应、7个异构化、30个自由基分解、16个自由基结合、7个芳构化和3个分子反应。计算过程采取试差法求取反应的动力学模型参数,该模型能够很好地预测MCH在20%~95%范围内的转化性能和关键组分选择性。另外,Pant等[44]还考察了MCH在负载K2CO3的铝酸钙催化剂上的蒸汽裂解情况。总的催化裂解反应速率可以用一阶反应速率方程表示,指数前因子为1.1×103m3/(kg·s),Ea=106.9 kJ/mol。无催化剂的MCH裂解的指数前因子和Ea分别为1.71×1011s-1和209.0 kJ/mol。建立的分子水平的动力学模型包含1个总反应和24个二次反应,能够很好地满足实验中得到的产物分布[45]。
从对MCH转化的整个反应过程的分析可看出,催化剂的结构性质和工艺条件对产物的选择性具有十分重要的影响。一方面,可以通过选取具有合适孔道结构、表面酸强度和酸类型的催化剂来控制产物分布;另一方面,通过控制反应的工艺参数也可以改变产物分布。催化裂化过程中氢转移反应是一个低活化能的放热过程,低温有利于氢转移反应的发生,对于产物中烯烃、芳烃和烷烃的产率影响明显。另外,增大反应空速,缩短反应物在催化剂上的停留时间也能减少氢转移反应[3]。
Quintana-Solórzano等[46]将单事件模型的概念引入到了环烷烃在平衡剂上的催化裂化反应,通过对693~753 K反应区间的裂化反应数据进行处理,回归得到了模型参数,模型建立时充分考虑到了环烷烃反应的化学特点。所建立的模型对于温度效应、不同烯烃烷烃比具有很好的预测能力。
5 现有研究的不足
通过对以上文献的分析可以清楚地看出,已有的文献报道采用的反应温度较低,低于典型的催化裂化反应温度(482~538 ℃)。如Santikunaporn等[11]和Raichle等[13]采用的反应温度范围为250~400 ℃。虽然Corma等[8]、Mostad等[33]和Sousa-Aguiar等[32]采用了稍高的反应温度,但整个研究工作也只是在一个固定温度下进行,忽略了操作温度对环烷烃转化的影响。
此外,以往的研究也很少考察催化剂和碳氢化合物的接触时间对环烷烃模型化合物总体转化的影响。在进行实验室研究中没有使用工业催化裂化反应时间,其范围是2~8 s[16]。微型活性测试装置中反应物与催化剂接触时间并不代表工业运行的反应条件,因此,不能提供预测工业生产催化剂性能的信息。
特别是对MCH分子水平的动力学模型建立以及分子筛的选择性开环等方面的研究不深入。另外,个别研究只考察了模型化合物的催化裂化总转化率,没有考虑产物的选择性,如苯和甲苯等芳香族化合物产物的产率未见报道,为确保燃料的质量和辛烷值水平,这些产物必不可少。
6 结语
MCH催化裂化催化剂、转化机理以及动力学模型的建立,对于认识多环环烷烃和环烷芳烃在催化裂化中的转化规律具有重要的借鉴意义,它是催化裂化技术开发的重要内容,有助于重质油轻质化和多产中间馏分。目前,从国内外的文献资料和专利申请可看出,相关研究的深度和广度还有待于进一步提高,尤其是对于多环环烷烃和环烷芳烃在催化裂化中转化规律的研究。环烷烃催化裂化催化剂的构效关系和催化裂化性能还需结合多种表征手段作进一步的研究。计算化学的广泛运用,可为催化剂的设计提供理论指导,避免催化剂开发的盲目性,节约时间和降低经济成本,加速催化剂开发的步伐。开发具有高活性、高稳定性、抗积碳性、选择性开环以及择形催化等多功能复合催化剂,不断优化配方和工艺条件,都将对重质油的开发利用具有十分重要的现实意义。
[1] Al-Sabawi M. Heterogeneous Kinetic Modeling of the Catalytic Cracking of Cycloparaffins[D]. Ontario:University of Western Ontario,2009.
[2] 陈俊武. 催化裂化工艺与工程[M]. 2版. 北京:中国石化出版社,2005:436.
[3] 高永灿,张久顺. 催化裂化过程中氢转移反应的研究[J]. 炼油设计,2000,30(11):34 - 38.
[4] Dewachtere N V,Santaella F,Forment G F. Application of a Single-Event Kinetic Model in the Simulation of an Industrial Riser Reactor for the Catalytic of Vacuum Gas Oil[J]. Chem Eng Sci,1999,54(15/16):3653 - 3660.
[5] 崔守业,许友好,程从礼,等. 正己烷在FAU和MFI型分子筛催化剂上的转化途径[J]. 石油学报:石油加工,2009,25(5):625 - 631.
[6] 唐津莲,许友好,汪燮卿,等. 四氢萘在分子筛催化剂上环烷环开环反应的研究[J]. 石油炼制与化工,2012,43(1):20 - 24.
[7] Watson B A,Klein M T,Harding R H. Catalytic Cracking of Alkylcyclohexanes:Modeling the Reaction Pathways and Mechanisms[J]. Int J Chem Kinet,1997,29(7):545 -560.
[8] Corma A,Mocholi F,Orchilles V,et al. Methylcyclohexane and Methylcyclohexene Cracking over Zeolite-Y Catalysts[J].Appl Catal,A,1990,67(2):307 - 324.
[9] Corma A,Agudo A L. Isomerization,Dehydrogenation and Cracking of Methylcyclohexane over HNaY Zeolites[J]. React Kinet Catal Lett,1981,16:253 - 257.
[10] Corma A,Martinez C,Melo F V,et al. A New Continuous Laboratory Reactor for the Study of Catalytic Cracking[J].Appl Catal,A,2002,232(1):247 - 263.
[11] Santikunaporn M,Alvarez W E,Resasco D E. Ring Contraction and Selective Ring Opening of Naphthenic Molecules for Octane Number Improvement[J]. Appl Catal,A,2007,325(1):175 - 187.
[12] Scofield C F,Benazzi E,Cauffriez H,et al. Methylcyclohexane Conversion to Light Olefins[J]. Braz J Chem Eng,1998,15(2):218 - 224.
[13] Raichle A,Traa Y,Weitkamp J. Preparation of a High-Quality Synthetic Steamcracker Feedstock from Methylcyclohexane on Acidic Zeolite H-ZSM-5:Influence of the Hydrogen Partial Pressure[J]. Appl Catal,B,2003,41(1):193 - 205.
[14] Cerqueira H S,Mihindou-Koumba P C,Magnoux P,et al.Methylcyclohexane Transformation over HFAU,HBEA,and HMFI Zeolites:Ⅰ. Reaction Scheme and Mechanisms[J].Ind Eng Chem Res,2001,40(4):1032 - 1041.
[15] Mihindou-Koumba P C,Cerqueira H S,Magnoux P,et al.Methylcyclohexane Transformation over HFAU,HBEA,and HMFI Zeolites:Ⅱ. Deactivation and Coke Formation[J]. Ind Eng Chem Res,2001,40(4):1042 - 1051.
[16] Al-Sabawi M,de Lasa H. Kinetic Modeling of Catalytic Conversion of Methylcyclohexane over USY Zeolites:Adsorption and Reaction Phenomena[J]. AIChE J,2009,55(6):1538 - 1558.
[17] Al-Sabawi M,de Lasa H. Influence of Zeolite Crystallite Size on Methyl-Cyclohexane Catalytic Conversion Products[J].Fuel,2012,96:511 - 523.
[18] Matias P,Lopes J M,Laforge S,et al. Methylcyclohexane Transformation over HMCM22 Zeolite:Mechanism and Location of the Reactions[J]. J Catal,2008,259(2):190 -202.
[19] Rabeharitsara A,Cerqueira H S,Magnoux P,et al. Trans-formation of Methylcyclohexane on a FCC Catalyst[J]. Braz J Chem Eng,2003,20(2):105 - 110.
[20] Garin F,Girard P,Maire G,et al. Isomerization of 2-Methylpentane and Ring Opening of Methylcyclopentane over Pt-Co/NaY Catalysts[J]. Appl Catal,A,1997,152(2):237 -247.
[21] Sedran U,de la Puente G. Conversion of Methylcyclopentane on Rare Earth Exchanged Y Zeolite FCC Catalysts[J]. Appl Catal,A,1996,144:147 - 158.
[22] Yin Changlong,Liu Chengguang. Transformation of Cycloparaffin over Zeolite-Supported Metal Catalyst for Improving the Octane Number of Gasoline[J]. Petrol Sci Technol,2005,23:1153 - 1161.
[23] Abbot J. Active Sites and Intermediates for Isomerization and Cracking of Cyclohexane on HY[J]. J Catal,1990,123(2):383 - 395.
[24] 袁帅,龙军,田辉平,等. 烃分子尺寸及其与扩散能垒关系的初步研究[J]. 石油学报:石油加工,2011,27(3):276 - 380.
[25] 袁帅,龙军,周涵,等. 芳烃、环烷烃分子在MFI和FAU分子筛中扩散行为的分子模拟[J]. 石油学报:石油加工,2011,27(4):508 - 515.
[26] Silva P V. Methylcyclohexane and n-Heptane Transformations over MCM-22 Zeolite[D]. Patrícia Matias:Université de Poitiers,2008.
[27] 张领辉,李再婷,许友好. 不同晶粒大小的ZSM-5分子筛催化剂的裂化反应差异[J]. 石油炼制与化工,1995,26(10):38 - 42.
[28] Borm R V,Reyniers M F,Martens J A,et al. Catalytic Cracking of Methylcyclohexane on FAU,MFI,and Bimodal Porous Materials:Influence of Acid Properties and Pore Topology[J]. Ind Eng Chem Res,2010,49(21):10486 -10495.
[29] 吴通好,许宁. MCM-22 族分子筛的结构及催化性能[J].化学通报,2004,67:1 - 21.
[30] Marques J P,Gener I,Lopes J M,et al. Methylcyclohexane Transformation over Dealuminated HBEA Samples:Mechanisms and Active Sites[J]. Appl Catal,A,2006,301(1):96 - 105.
[31] Cerqueira H S,Magnoux P,Martin D,et al. Effect of Contact Time on the Nature and Location of Coke During Methylcyclohexane Transformation over a USHY Zeolite[J]. Stud Surf Sci Catal,1999,126:105 - 112.
[32] Sousa-Aguiar E F,Mota C J A,Murta V M L,et al. Catalytic Cracking of Decalin Isomers over REHY Zeolites with Different Crystallite Sizes[J]. J Mol Catal A:Chem,1996,104(3):267 - 271.
[33] Mostad H B,Riis T U,Ellestad O H. Catalytic Cracking of Naphthenes and Naphtheno-Aromatics in Fixed Bed Micro Reactors[J]. Appl Catal,1990,63(1):345 - 364.
[34] Mostad H B,Riis T U,Ellestad O H. Shape Selectivity in YZeolites:Catalytic Cracking of Decalin-Isomers in Fixed Bed Micro Reactors[J]. Appl Catal,1990,58(1):105 - 117.
[35] Tang Jinlian,Xu Youhao,Wang Xieqing. Naphthenic Ring Opening of Perhydrophenanthrene over Zeolite Catalysts[J]. J Fuel Chem Technol,2012,40(6):721 - 726.
[36] Al-Sabawi M,de Lasa H. Modeling Thermal and Catalytic Conversion of Decalin Under Industrial FCC Operating Conditions[J]. Chem Eng Sci,2010,65(2):626 - 644.
[37] Castaño P,Arandes J M,Olazar M,et al. Effect of Hydrogen on the Cracking Mechanisms of Cycloalkanes over Zeolites[J]. Catal Today,2010,150(3/4):363 - 367.
[38] Moraes R,Thomas K,Thomas S,et al. Ring Opening of Decalin and Methylcyclohexane over Alumina-Based Monofunctional WO3/Al2O3and Ir/Al2O3Catalysts[J]. J Catal,2012,286:62 - 77.
[39] Rabl S,Haas A,Santia D,et al. Ring Opening of cis-Decalin on bifunctional Ir/- and Pt/La-X Zeolite Catalysts[J]. Appl Catal,A,2011,400(1/2):131 - 141.
[40] Kubička D,Salmi T,Tiitta M,et al. Ring-Opening of Decalin: Kinetic Modelling[J]. Fuel,2009,88:366 - 373.
[41] Nylén U,Sassu L,Melis S,et al. Catalytic Ring Opening of Naphthenic Structures:Ⅰ. From Laboratory Catalyst Screening via Pilot Unit Testing to Industrial Application for Upgrading LCO into a High-Quality Diesel-Blending Component[J].Appl Catal,A,2006,299:1 - 13.
[42] Sirjean B,Glaude P A,Ruiz-Lopèz M F,et al. Theoretical Kinetic Study of Thermal Unimolecular Decomposition of Cyclic Alkyl Radicals[J]. J Phys Chem,2008,112(46):11598 - 11610.
[43] Singh O V,Pant K K. Mechanistic Modelling of the Catalytic Pyrolysis of Methylcyclohexane[J]. Can J Chem Eng,2003,81(5):981 - 992.
[44] Pant K K,Kunzru D. Catalytic Pyrolysis of Methylcyclohexane:Kinetics and Modeling[J]. Chem Eng J,1998,70(1):47 - 54.
[45] Pant K K,Kunzru D. Pyrolysis of Methylcyclohexane:Kinetics and Modeling[J]. Chem Eng J,1997,67(2):123 - 129.
[46] Quintana-Solórzano R, Thybaut J W, Marin G B. A Single-Event Microkinetic Analysis of the Catalytic Cracking of(Cyclo) Alkanes on an Equilibrium Catalyst in the Absence of Coke Formation[J]. Chem Eng Sci,2007,62(18/20):5033 - 5038.
猜你喜欢
石油炼制与化工(2022年6期)2022-06-21
燃料化学学报(2022年5期)2022-05-30
煤炭转化(2020年2期)2020-04-24
石油石化绿色低碳(2019年6期)2019-01-14
石油石化绿色低碳(2019年6期)2019-01-14
润滑油(2016年4期)2016-11-04
当代化工研究(2016年6期)2016-03-20
合成化学(2015年4期)2016-01-17
化工进展(2015年6期)2015-11-13
海军航空大学学报(2015年1期)2015-11-11
