
两性离子催化剂的研究进展
2013-10-11 08:35聂万丽MaximBorzov
化工进展 2013年10期
聂万丽,曹 蓉,Maxim V Borzov
(1乐山师范学院化学学院,四川 乐山 614000;2西北大学化学与材料科学学院,陕西 西安 710069)
在过去的三十年里,有关茂金属催化剂在聚烯烃工业中表现的研究备受关注。茂金属催化剂在催化烯烃均相聚合反应中所表现出的高催化活性、单一活性中心和高立构规整性使它成为20世纪80年代金属有机化学领域的研究热点之一。
有关茂金属催化剂体系催化烯烃聚合的过程相对较复杂[1-4]。已被广泛认可的机理为:催化剂前体(中性的第四副族茂金属二氯化物及二甲基衍生物)与一个作为助催化剂的强路易斯酸反应,得到一个中心金属离子具有 14电子结构的阳离子活性反应中心。在催化反应过程中,保证这一阳离子反应活性中心不受到反应体系中碱性杂质或其它物质的影响是维持催化剂寿命的关键因素。工业生产中为了提高聚合活性,反应中通常需要使用大量的助催化剂甲基铝氧烷MAO(Al∶M = 100~10000∶1),它是一种由三甲基铝部分水解得到的混合物,因此以茂金属为催化剂的烯烃生产成本大大提高。针对这一缺陷,近几年有关新型茂金属催化剂及非茂有机金属催化剂体系的研究工作十分活跃。
针对茂金属配合物催化烯烃聚合反应的机理特征,在催化反应过程中,为了保证金属阳离子活性中心不受到反阴离子的影响,化学家们提出了一种两性离子茂金属催化剂的设计理念。即将阴离子固定在配合物的配体上,在远离金属中心的某些区域共价地鳌合阴离子。这种强制的物理隔离作用可减少阴阳离子对之间相互猝灭的机会,延长催化剂的催化寿命。
相对于传统的双组分催化剂,两性离子茂金属催化剂至少可预见两大优点[5]:第一,从工业化的工艺流程角度而言,两性离子催化剂是结构明确的单组分催化剂,无需助催化剂活化;第二,此类催化剂在进行烯烃聚合反应的烃类溶剂中具有更好的溶解性。虽然两性离子茂金属催化剂的合成较为困难,但是敢于挑战的化学家依旧对两性离子配合物的研究进行了大量探索。
在两性离子催化剂的设计上,科学家们按照阴离子在配体上所连接区域的不同将其分为三类[5](图1):Girdle型即阴离子被固定在配合物腰部配体的烷基基团上;Ring型即阴离子被固定在环戊二烯基环上;Birdge型即阴离子被固定在配体的桥链上。Birdge型两性离子虽然在设计上可以利用很多柄型茂金属配合物的丰富结构,而且还可使阴阳离子电荷被完全分离,但是至今尚未成功合成得到此类两性离子茂金属配合物。
本文主要针对目前文献中报道的 Girdle型和Ring型两种两性离子茂金属催化剂的结构特征和反应活性进行归纳,并且针对目前受关注的具有两性离子特征的非茂有机金属催化剂的研究也进行了总结。希望本文能够为国内同行提供关于两性离子催化剂研究的有益的参考价值。
1 两性离子茂金属配合物的研究进展
1.1 Girdle型两性离子茂金属配合物
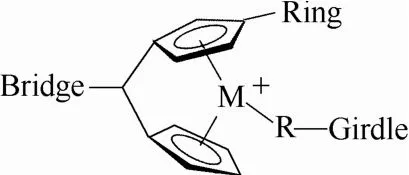
图1 两性离子茂金属配合物的结构设计
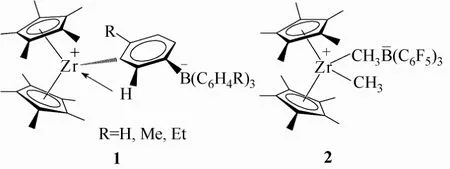
图2 Hlatky,Turner等及Marks等报道的Girdle型配合物
1989年,Hlatky 和Turner等[6]使用[Cp*2ZrMe2]和[Bu3NH]+[B(C6H4R)]–反应,得到了首例两性离子配合物1,见图2。配合物的结构表明芳环邻位上的C—H键与金属的相互作用可以使中心阳离子更加稳定,使Zr金属中心与苯环碳之间形成更强的化学键。两性离子配合物1所表现的优良聚烯烃催化活性,激励了其他科学家开始探索新的Girdle型两性离子催化剂体系。
1991年,Marks等[7-8]将B(C6F5)3作为一个强路易斯酸引入到二茂锆衍生物中,生成了Girdle型配合物 2,见图2。Girdle型配合物 2中的 B(C6F5)3相比配合物1中芳基硼烷的配位能力更弱,更有利于两性离子配合物的稳定存在。在Marks的研究之后,有关大位阻弱配位路易斯酸的研究主要集中于氟代芳基硼烷或氟代芳基铝。
1992年,Lapointe及其合作者[9]报道了一种具有限定几何构型特征的两性离子配合物3,见图3,它在催化乙烯、α-烯烃和MMA(甲基丙烯酸甲酯)嵌段共聚中都表现出高催化活性。其后在1997年和2001年Marks和Royo小组分别研究了两类具有限定几何构型的两性离子配合物。Marks及其合作者[10-11]所报道的限定几何构型两性离子配合物 4,见图3,在催化乙烯和丙烯聚合中都表现出达到工业要求的高反应活性,所得产物具有较高的分子量和热转变温度,但产物聚合度分布范围变宽。而Royo等[11-12]报道的是一种具有双硅-氨基桥联的两性离子茂金属配合物5(图3),该类配合物在催化长链的α-烯烃聚合时拥有良好的活性。
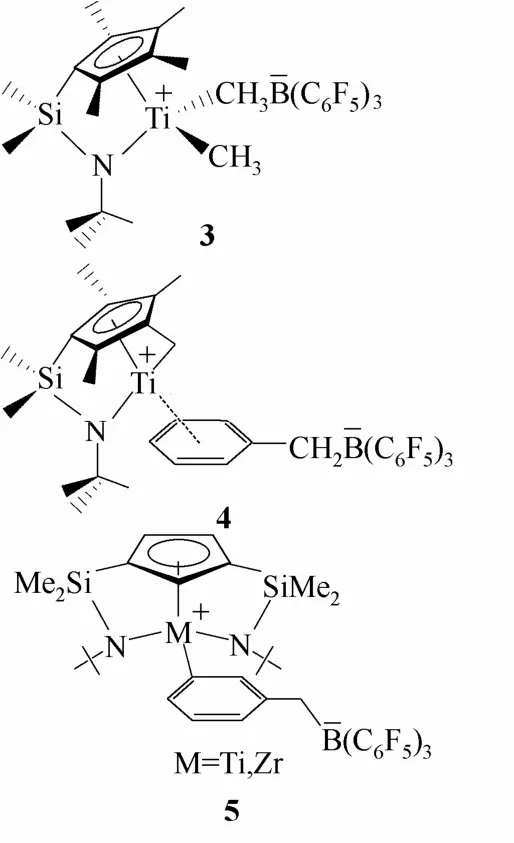
图3 具有限定几何构型的Girdle型配合物
1995 年,Erker小组[14-16]对[Cp2Zr(η4-丁二烯)]和B(C6F5)3反应所生成的两性离子茂金属配合物6(图4)的结构、催化烯烃聚合时的反应活性以及反应机理进行了详细的研究。配合物6中芳环上的C—F键和Zr金属中心间存在的相互作用使得金属阳离子可以稳定存在。基于这种弱的给电子效应,该结构被认为是Girdle型两性离子茂金属配合物的普遍特征。Erker小组利用NMR研究了配合物6在溶液中的动力学行为,从其氟谱图中观察到这类配合物的M—F键在溶液中容易解离。因此,该配合物在溶液中与其配位不饱和的异构体相互平衡,并表现出高的聚烯烃催化活性。
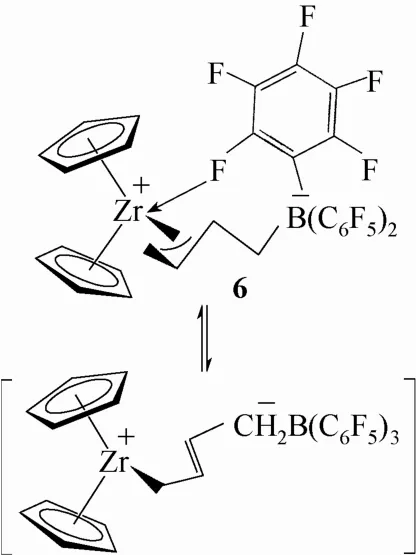
图4 Erker报道的Girdle型配合物
Erker小组[14]通过研究该类两性离子配合物的反应机理发现,Girdle型两性离子配合物所具有的两性特征在催化条件下维持的时间很短暂,经过多次烯烃插入反应后转变成了一个常规的、非两性配合物而使得反应活性消失。这种失活现象也通过其它一些反应研究得到证实,比如Bochmann等[17]于1998年在研究双茂金属配合物 Cp2ZrMe2与Al(C6F5)3反应时,发现产物并非稳定的两性离子配合物,而是一个非两性、活性极低的配合物 7,见图5。2004年,Fenwick等[9,18]在研究一类含芳基的单茂双甲基钛配合物与B(C6F5)3的反应中也同样得到了一个 C6F5基团转移后形成的非两性的低活性配合物8,见图5。
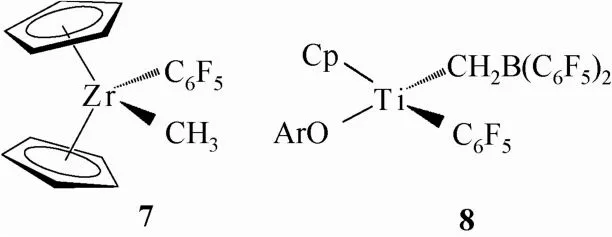
图5 失活的非两性产物
2001—2002 年,Chen等[19-21]报道了一类使用强路易斯酸 Al(C6F5)3合成的两性离子茂金属配合物9,10,11,见图6。其中配合物9在催化烯烃聚合中具有良好的链转移功能,可作为催化MMA间规聚合的高活性催化剂。与之形成对比的是,使用B(C6F5)3代替 Al(C6F5)3并不能得到类似的目标产物,只生成一些混合物。配合物10,11均是经过双活化反应得到的具有双阳离子中心特征的两性离子配合物。这种双活化结构可以降低聚合反应引发和链增长的能垒而使反应活性增强。在催化乙烯及1-辛烯的聚合反应中,该类双阳离子中心配合物较相应的含路易斯酸B(C6F5)3的配合物从初始的聚合物放热到催化效率都有了大幅提高,并且产物分子量有了明显提高。但是若使用1 mol的Al(C6F5)3反应,所得到的相应的单阳离子配合物的活性却比相应的硼烷配合物低。
1.2 Ring型两性离子茂金属配合物
Ring型两性离子茂金属催化剂有两种类型:一种为B(C6F5)3与茂环间通过一个短碳链相连;另一种为B(C6F5)3直接连接在茂环上。
1.2.1 阴离子与茂环间通过一个短碳链相连的Ring型两性离子茂金属配合物
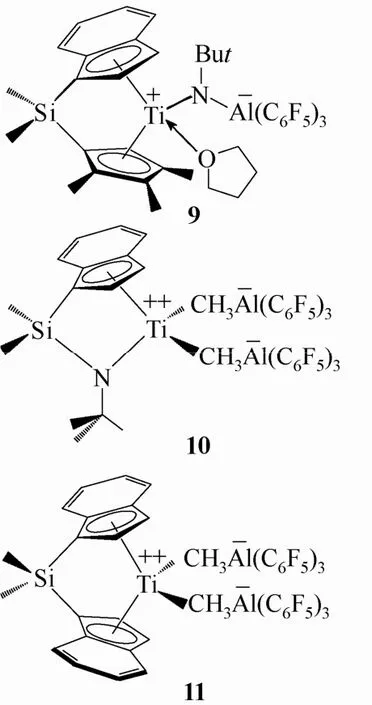
图6 含有Al(C6F5)3的Girdle型配合物
将B(C6F5)3与茂环间通过一个短碳链相连的方式一般都是通过B(C6F5)3与茂环侧链上不饱和基团进行的反应。
1997年,Marks等[22-23]报道了一种B(C6F5)3与茂环间隔一个碳原子的Ring型两性离子配合物12,见图7,它是由一种被称为Tuck-in式的二茂锆配合物与硼烷反应得到的,已被证明是一种高活性的单组分乙烯聚合催化剂。配合物12的结构是热力学稳定的,其动力学稳定结构也具有与Girdle型两性离子配合物类似的Zr··F—C相互作用。
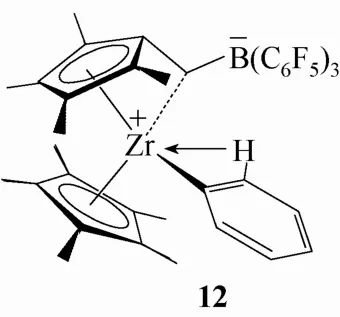
图7 Ring型“Cp-C-B”两性离子配合物
通过大量的实验科学家们发现,硼烷与茂环之间的碳链长度对两性离子的稳定性尤为重要,当间隔 3个碳原子时配合物会发生重排而失去两性特征[24-25]。
1.2.2 阴离子直接连在茂环上的 Ring型两性离子茂金属配合物
将 B(C6F5)3直接连接在茂环上的方法有很多种。1995年Bochmann和他的合作者们[26]在配合物合成前通过环戊二烯的锂盐和B(C6F5)3反应将硼烷引入到配体的茂环上,然后与相应的金属盐反应成功得到了Ring型两性离子配合物13,见图8。该配合物的金属阳离子中心与Al(CH3)3络合,因此阴阳离子电荷能很好地被分隔开来。如果没有烷基铝的参与则会形成与Girdle型两性离子配合物结构类似M··F—C 键相互作用。
1996年,Erker等[27]报道了一种二茂锆两性离子配合物14,见图8。在配合物的合成过程中硼烷没有进攻二烯体的σ轨道形成Girdle型两性离子化合物,而是很意外地进攻了其中一个茂环,环上质子转移至乙烯基碳与中心金属锆之间的化学键上而生成Ring型两性离子配合物。
Piers[5]在研究炔类配合物[Cp2Zr(η2-EtC≡CEt)(PMe3)]和HB(C6F5)2的反应时也发现了类似的反应现象。他们通过NMR分析推测反应得到的产物可能是具有两性离子特征的配合物15,见图8。反应没有得到Girdle型两性离子化合物的原因可能是由于 Zr—C键上的空间位阻较大而阻止了硼烷的进攻,硼烷对茂环的亲电进攻形成了Ring型产物。
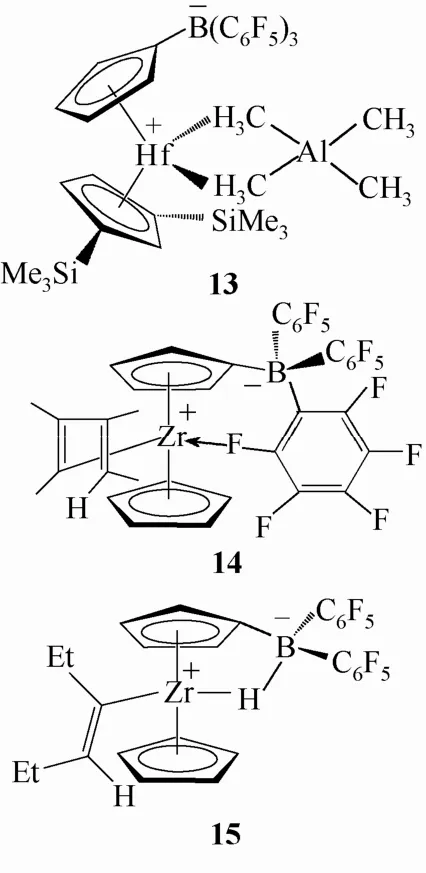
图8 Ring型“Cp-B”两性离子配合物
Ring型茂金属两性离子配合物不仅在催化烯烃聚合反应中表现出高活性,而且在其它类型反应中的表现同样受到科学家们的关注。2001年,Burlakov等[28]报道了一类由氧原子桥联的双核两性离子配合物16,17,见图9。此类配合物在催化α-烯烃聚合反应中活性不佳,但是它们在催化ε-CL的开环聚合中表现出良好的活性。在催化THF开环聚合的反应研究中,配合物16活性较好而配合物17的活性较低。同时,其单核配合物还可以与丙酮反应生成两性离子配合物18[29],见图9。这是第一类被报道的含羰基的两性离子配合物。羰基氧原子与金属中心配位,C=O键被大大削弱,因此推测此类两性茂金属化合物可作为一种新型的醛、酮亲核反应催化剂。
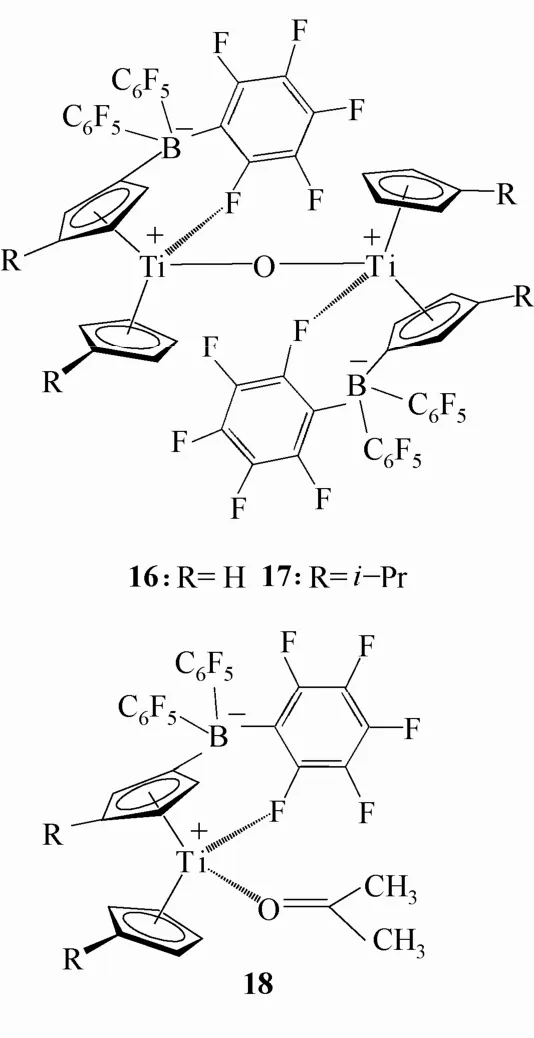
图9 Burlakov等报道的Ring型两性离子配合物
2 非茂两性离子前过渡金属配合物的研究进展
尽管茂金属催化剂已经发展了几十年并取得了一定的成果,然而其中所存在的很多缺点促使着人们不断探索其它类型的催化剂。科学家们尝试利用其它含杂原子的基团代替茂金属配合物中的一个或两个茂环合成非茂金属催化剂。目前,这一研究方向已成为一大热点。针对非茂两性离子催化剂的研究也同样受到重视。
2010年,Cabrera等[30]报道了这样一种含有多齿配体的两性离子配合物19(图10),具有路易斯酸性的B(C6F5)3被固定在远离阳离子金属中心的氰基官能团上,通过共轭效应的传递使金属中心上的电荷密度降低,增强了催化反应活性。该化合物在催化乙烯聚合的研究中表现出良好的活性,且MAO用量大大下降。
2012年徐莹莹[31]报道了一类含咪唑杂环的两性离子配合物20(图10),实验已证实该两性离子配合物较其相应的中性配合物前体的反应活性明显提高,在催化交叉去氢偶联反应和活化CO2等小分子的反应中均表现出了独特的优越性。
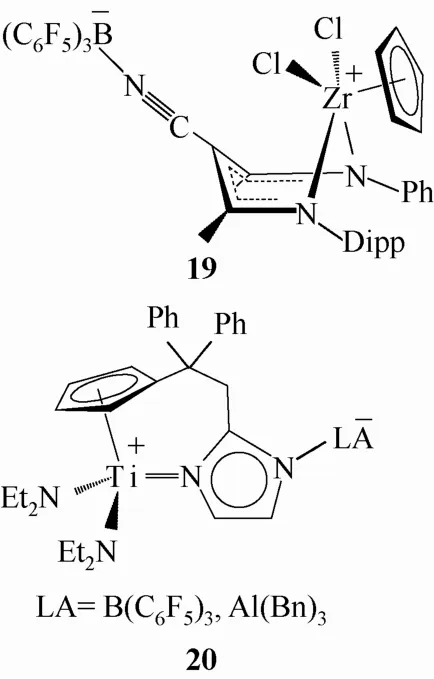
图10 含B(C6F5)3的非茂两性离子配合物
非茂两性离子配合物中当然也少不了Al(C6F5)3的身影。2006年,Tsurugi和 Mashima[32]报道了一种使用 Al(C6F5)3制备的两性离子非茂金属配合物21,见图11。在该结构中,苄基铝酸盐阴离子和中心金属Zr之间的相互作用更强,然而相应的B(C6F5)3衍生物却非常不稳定,无法分离得到。配合物 21在催化 1-己烯的聚合反应中表现出单一活性中心的催化特征,所得聚合产物也具有较窄的分子量分布。
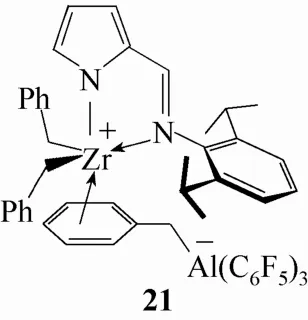
图11 含Al(C6F5)3的非茂两性离子配合物
3 非茂两性离子后过渡金属配合物的研究进展
有关非茂两性离子配合物的研究除了以上例举的前过渡金属配合物以外,具有两性离子特征的后过渡金属非茂配合物的研究也有大量的文献报道。
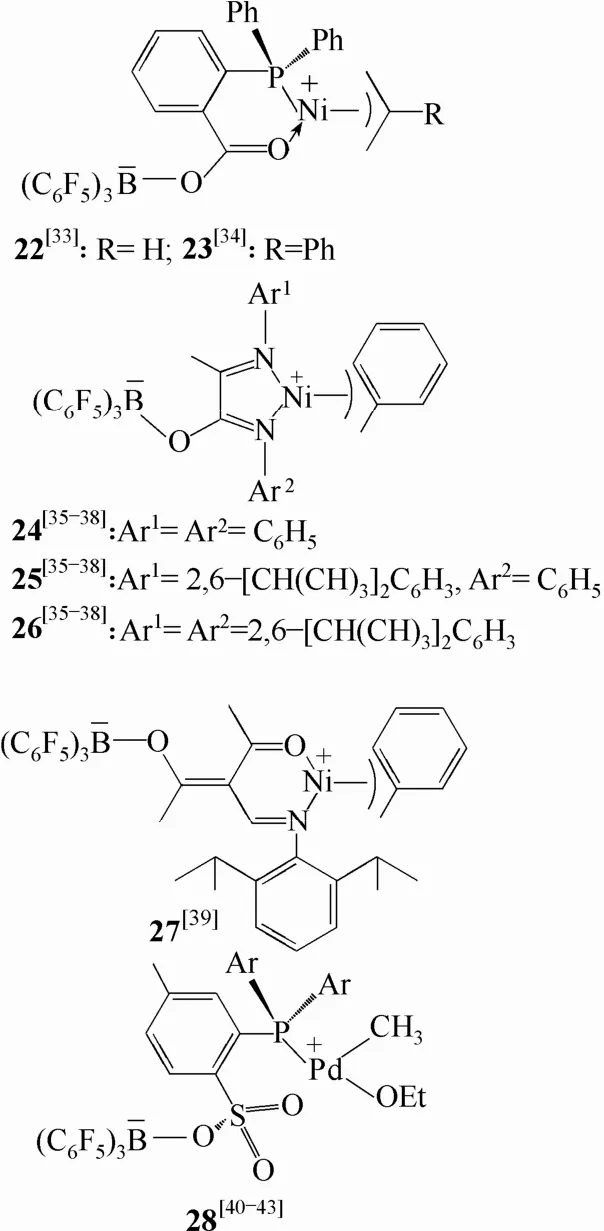
图12 非茂两性离子后过渡金属配合物
图12中列举出了近几年报道的具有代表性的后过渡金属两性离子配合物[33-43]。它们由含有共轭杂原子配体的中性后过渡金属配合物与B(C6F5)3作用后转化形成,并且均对乙烯聚合表现出良好的催化活性。
2009年,Onishi等[44-45]报道了一系列含有nbd或tfb配体的Rh两性离子配合物29~33,见图13。他们利用 NaBPh4与中性的氯化铑配合物反应,可得到相应的两性离子配合物。它们较相应的中性Rh配合物在苯乙炔及其衍生物的聚合反应中表现出更好的催化活性,而且它们的固态很稳定,甚至可以暴露于空气中,只是在溶液中易分解。
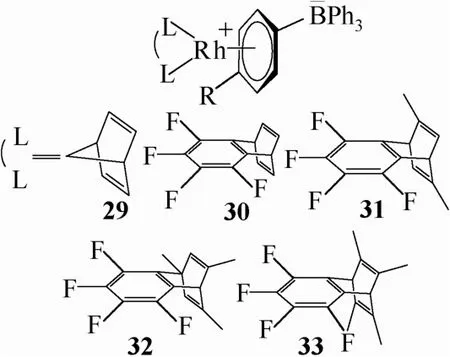
图13 Rh两性离子配合物
后过渡金属两性离子配合物不仅是良好的烯烃聚合催化剂,同时它们还可以催化多种类型的小分子反应。早在1990年,Amer等[46]就发现图13中Rh两性离子配合物在CO、H2存在下可以与多种烯烃、炔烃发生酰化反应或硅氢化反应,且反应条件温和,反应过程具有高度的区域选择性。其它的相关研究亦表明,后过渡金属两性离子配合物可催化CO与乙烯单体的共聚[47]以及高活性的催化苯乙烯硅氢加成及加氢酰化[48-49]等多种反应。
4 两性离子催化剂的研究展望
总的来说,有关两性离子配合物的研究已经取得了很多可喜的成果。虽然两性离子配合物在烯烃聚合、开环聚合、小分子活化等多个领域中都表现出良好的反应活性,但是目前有关两性离子配合物的合成仍较为困难,并且全氟芳基硼烷或全氟芳基铝的价格也较高,这些缺点使它的发展受到了限制。现阶段,国内科学家们对此领域的研究也较少,目前探索简便高效的新型两性离子配合物的研究课题有待科学家们进一步解决。有关两性离子催化剂的应用还有很多未知的领域有待开发,尤其是在小分子的活化、催化领域具有潜在的应用前景。有理由相信,未来两性离子催化剂的发展必将绽放耀眼的光芒。
[1] Brintzinger H H,Fischer D,Mulhaupt R,et al. Stereospecific olefin polymerization with chiral metallocene catalysts[J].Angew. Chem.,1995,34(11):1143-1170.
[2] Bochmann M. Cationic group 4 metallocene complexes and their role in polymerisation catalysis:The chemistry of well defined Ziegler catalysts[J] .J. Chem. Soc. Dalton Trans.,1996(3):255-270.
[3] Marks T J. Surface-bound metal hydrocarbyls. Organometallic connections between heterogeneous and homogeneous catalysis[J].Acc Chem. Res.,1992,25(2):57-65.
[4] Jordan R F. Chemistry of cationic dicyclopentadienyl group 4 metal-alky I complexes[J].Adv. Organomet. Chem.,1991,32:325-387.
[5] Piers W E. Zwitterionic metallocenes[J].Chem. Eur. J.,1998,4(1):13-18.
[6] Hlatky G G,Turner H W,Eckman R R. Ionic,base-free zirconocene catalysts for ethylene polymerization[J].J. Am. Chem. Soc.,1989,111(7):2728-2729.
[7] Massey A G,Park A J. Perfluorophenyl derivatives of the elements:Ⅰ. Tris(pentafluorophenyl)boron[J].J. Organomet. Chem.,1964:2(3):245-250.
[8] Yang X,Stern C L,Marks T J. Cation-like homogeneous olefin polymerization catalysts based upon zirconocene alkyls and tris(pentafluorophenyl)borane[J].J. Am. Chem. Soc.,1991,113(9):3623-3625.
[9] Chen E Y X. Cocatalysts for metal-catalyzed olefin polymerization:Activators ,activation processes ,and structure−activity relationships[J].Chem. Rev.,2000,100(4):1391-1434.
[10] Chen Y X ,Marks T J. “Constrained geometry” dialkyl catalysts.Efficient syntheses,C—H bond activation chemistry,monomer-dimer equilibration ,andα-olefin polymerization catalysis[J].Organometallics,1997,16(16):3649-3657.
[11] Chen Y X,Fu P F,Stern C L,et al. A novel phenolate “constrained geometry” catalyst system. Efficient synthesis ,structural characterization ,and α-olefin polymerization catalysis[J].Organometallics,1997,16(26):5958-5963.
[12] Cano J,Royo P,Lanfranchi M,et al. A new type of doubly silylamido-bridged cyclopentadienyl group 4 metal complexes[J].Angew. Chem. Int. Ed.,2001,40(13):2495-2497.
[13] Bochmann M. The chemistry of catalyst activation:The case of group 4 polymerization catalysts[J].Organometallics,2010,29(21):4711-4740.
[14] Temme B,Erker G,Karl J,et al. Reaction of (Butadiene)zirconocene with tris(pentafluorophenyl)borane——A novel way of generating methylalumoxane-free homogeneous Ziegler-Type catalysts[J].Angew. Chem. Int. Ed. Engl.,1995,34(16):1755-1757.
[15] Temme B,Karl J,Erker G. Observing a homogeneous Ziegler catalyst precursor at work:Insertion reactions into the zirconium–carbon bond of the (butadiene) ZrCp2/B(C6F5)3addition product[J].Chem. Eur. J.,1996,2(8):919-924.
[16] Karl J,Erker G,Fröhlich R. UV resonance raman ground and excited state studies of amide and peptide isomerization dynamics[J].J. Am.Chem. Soc.,1997,119(6):1116-1120.
[17] Bochmann M,Sarsfield M J. Reaction of AlR3with[CPh3][B(C6F5)4]:Facile degradation of [B(C6F5)4]−by transient“[AlR2]+”[J].Organometallic,1998,17(26):5908-5912.
[18] Fenwick A E,Phomphrai K,Thorn M G,et al. Formation of neutral and cationic methyl derivatives of titanium containing cyclopentadienyl and aryloxide ancillary ligation[J].Organometallics.2004,23(9):2146-2156.
[19] Jin J,Chen E Y X. Chiral ansa-titanocene imido complexes:Novel synthesis and effective initiator for syndiospecific polymerization of MMA[J].Organometallics. 2002,21(1):13-15.
[20] Chen E Y X,Kruper W J,Roof G,et al. “Double activation” of constrained geometry and ansa-metallocene group 4 metal dialkyls:Synthesis,structure,and olefin polymerization study of mono and dicationic aluminate complexes[J].J. Am. Chem. Soc.,2001,123(4):745-746.
[21] Chen E Y X. Coordination polymerization of polar vinyl monomers by single-site metal catalysts[J].Chem. Rev.,2009,109(11):5157-5214.
[22] Schock L E,Brock C P,Marks T J,et al. Intramolecular thermolytic carbon-hydrogen activation processes. Solid-state structural characterization of a mononuclear .eta.6-Me4C5CH2zirconium complex and a mechanistic study of its formation from(Me5C5)2Zr(C6H5)2[J].Organometallics,1987,6(2):232-241.
[23] Yang X,Stern C L,Marks T J. Cationic zirconocene olefin polymerization catalysts based on the organo-Lewis acid tris(pentafluorophenyl)borane. A synthetic,structural,solution dynamic,and polymerization catalytic study[J].J. Am. Chem. Soc.,1994,116(22):10015-10031.
[24] Spence R E von H,Piers W E. One-component group 4 homogeneous Ziegler-Natta olefin polymerization catalysts:Hydroboration of zirconium bisalkyls with pendant 2-propenyl groups using[(C6F5)2BH]2[J].Organometallics,1995,14(10):4617-4624.
[25] Spence R E von H,Parks D J,Piers W E,et al. Konkurrierende reaktionswege bei der reaktion von bis(pentafluorphenyl)boran mit bis(η5-cyclopentadienyl) dimethylzirconium: Methan-eliminierung oder methyl- hydrid-austausch und ein beispiel für fünffach koordinierten kohlenstoff[J].Angew. Chem.,1995,107(11):1337-1340.
[26] Bochmann M,Lancaster S J,Robinson O B,et al. Anionic and zwitterionic metallocene complexes derived from novel boratocyclopentadienyl ligands[J].J. Chem. Soc.Chem.Commun.,1995,20:2081-2082.
[27] Ruwwe J,Erker G,Fröhlich R. Formation of a zirconocene–betaine system by electrophilic substitution with B(C6F5)3at a Cyclopentadienyl Ligand[J].Angew. Chem. Int. Ed. Engl.,1996,35(1):80-82.
[28] Burlakov V V,Arndt P,Baumann W,et al. Synthesis and X-ray crystal structure determination of new zwitterionic complexes of titanocene[J].Organometallics,2001,20(19):4072-4079.
[29] Burlakov V V,Letov A V,Arndt P,et al. Zwitterionic titanoxanes{Cp[η5-C5H4B(C6F5)3]Ti}2O and {(η5-iPrC5H4)[η5-1,3-iPrC5H3B(C6F5)3]Ti}2O as catalysts for cationic ring-opening polymerization[J].J. Mol. Catal. A:Chem.,2003,200(1):63-67.
[30] Cabrera,Alan R,Schneider Y,Valderrama M,et al. Synthesis of a[(π-Cyano-nacnac)Cp]zirconium complex and its remote activation for ethylene polymerization[J].Organometallics,2010,29(22):6104-6110.
[31] 徐莹莹,Borzov M V. 2-甲基咪唑类侧链桥联型茂金属配合物的合成、反应性及催化活性研究[D]. 西安:西北大学化学与材料化学学院,2012.
[32] Tsurugi H,Mashima K. Preparation and characterization of a zwitterionic (iminopyrrolyl)zirconium complex with benzylaluminate anion and its catalytic performance for 1-hexene polymerization[J].Organometallics,2006,25(22):5210-5212.
[33] Komon Z J A,Bu X,Bazan G C. Synthesis of butene - ethylene and hexane - butene - ethylene copolymers from ethyleneviatandem action of well-defined homogeneous catalysts[J].J. Am. Chem. Soc.,2000,122(8):1830-1831.
[34] Komon Z J A,Bu X,Bazan G C. Synthesis,characterization,and ethylene oligomerization action of [(C6H5)2PC6H4C(O—B(C6F5)3)O—K2P,O]Ni(η3-CH2C6H5)[J].J. Am. Chem. Soc.,2000,122(49):12379-12380.
[35] Lee B Y,Bazan G C,Vela J,et al. α-Iminocarboxamidato−nickel(Ⅱ)ethylene polymerization catalysts[J].J. Am. Chem. Soc.,2001,123(22):5352-5353.
[36] Rojas R S,Wasilke J C,Wu G,et al. α-Iminocarboxamide nickel complexes:Synthesis and uses in ethylene polymerization[J].Organometallics,2005,24(23):5644-5653.
[37] Diamanti S J,Ghosh P,Shimizu F,et al. Ethylene homopolymerization and copolymerization with functionalized 5-Norbornen-2-yl monomers by a novel nickel catalyst system[J].Macromolecules,2003,36(26):9731-9735.
[38] Boardman B M ,Bazan G C. α-Iminocarboxamidato nickel complexes[J].Acc. Chem. Res.,2009,42(10):1597-1606.
[39] Chen Y,Wu G,Bazan G C. Remote activation of nickel catalysts for ethylene oligomerization[J].Angew. Chem. Int. Ed.,2005,44(7):1108-1112.
[40] Berkefeld A,Mecking S. Coordination copolymerization of polar vinyl monomers H2C=CHX. [J]Angew. Chem. Int. Ed.,2008,47(14):2538-2542.
[41] Noda S,Nakamura A,Kochi T,et al. Mechanistic studies on the formation of linear polyethylene chain catalyzed by palladium phosphine−sulfonate complexes : Experiment and theoretical studies[J].J. Am. Chem. Soc.,2009,131(39):14088-14100.
[42] Cai Z,Shen Z,Zhou X,et al. Enhancement of chain growth and chain transfer rates in ethylene polymerization by (phosphine-sulfonate)PdMe catalysts by binding of B(C6F5)3to the sulfonate group[J].Catal.,2012,2(6):1187-1195.
[43] Kishimoto Y,Itou M,Miyatake T,et al. Polymerization of monosubstituted acetylenes with a zwitterionic rhodium(Ⅰ) complex,Rh+(2,5-norbornadiene) [.eta.6-C6H5)B−(C6H5)3][J].Macromolecules,1995,28(19):6662-6666.
[44] Nakamura A,Ito S,Nozaki K. Coordination-insertion copolymerization of fundamental polar monomers[J].Chem. Rev.,2009,109(11):5215-5244.
[45] Onishi N,Shiotsuki M,Sanda F,et al. Polymerization of phenylacetylenes with rhodium zwitterionic complexes:Enhanced catalytic activity by π-acidic diene ligands[J].Macromolecules,2009,42(12):4071-4076.
[46] Amer I,Alper H. Zwitterionic rhodium complexes as catalysts for the hydroformylation of olefins[J].J. Am. Chem. Soc.,1990,112(9):3674-3676.
[47] Betley T A,Peters J C. Zwitterionic relatives to the classic[(P–P)Rh(solv)2]+ions:Neutral catalysts active for H=E bond additions to olefins (E=C,Si,B)[J].Angew. Chem. Int. Ed.,2003,42(21):2385-2389.
[48] Lu C C,Peters J C. Catalytic copolymerization of CO and ethylene with a charge neutral palladium(Ⅱ) zwitterion[J].J. Am. Chem. Soc.,2002,124(19):5272-5273.
[49] Stradiotto M,Hesp K D,Lundgren R J. Zwitterionische verwandte kationischer platinmetallkomplexe: Anwendungen in der stöchiometrischen und katalytischen σ-bindungsaktivierung[J].Angew.Chem.,2010,122(3):504-523.
猜你喜欢
云南化工(2021年8期)2021-12-21
中国石油石化(2021年9期)2021-07-17
西南石油大学学报(自然科学版)(2018年6期)2018-12-26
当代化工研究(2016年2期)2016-03-20
中国洗涤用品工业(2015年7期)2015-02-28
现代农业(2015年3期)2015-02-28
应用化工(2014年1期)2014-08-16
应用化工(2014年12期)2014-08-16
无机化学学报(2014年9期)2014-02-28
郑州大学学报(理学版)(2012年4期)2012-03-25
