
维药尿通卡克乃其片中甘草浸膏的色谱鉴别方法研究△
2013-09-11 11:33:24祝莉莎赵荣梅伊布拉音艾尔西丁
中国民族医药杂志 2013年4期
祝莉莎 赵荣梅 王 毅 高 勇 伊布拉音·艾尔西丁
(1.新疆维吾尔自治区食品药品检验所,新疆 乌鲁木齐 830004;2.新疆喀什地区药品检验所,新疆 喀什 844000)
尿通卡克乃其片为维吾尔医常用成方制剂,由锦灯笼、黄瓜子、血竭、西黄耆胶、阿拉伯胶、巴旦仁、甘草浸膏等10味药材及辅料组成。具止痛、利尿等功效,临床用于尿通、尿不尽、尿血、尿道流脓等。该品种收载于《卫生部药品标准-维吾尔药分册》(1998年版);甘草浸膏系国家中药材资源保护品种甘草经加工制成的浸膏,在该制剂中为辅助药用成分。本文就该制剂中的甘草浸膏药味特征性成分甘草酸和甘草次酸的色谱鉴别方法进行了系统研究,以期为尿通卡克乃其片的内在质量控制提供依据。
1 实验材料
1.1 仪器:岛津 SPD-10AVP紫外检测器LC-10AT泵、岛津LC-2010A高效液相色谱仪和LC-2010AHT紫外检测器、Agilent1100高效液相色谱仪(二极管阵列检测器,美国安捷伦科技),KLZ-UP超纯水仪(台湾艾柯成都康宁实验专用纯水设备厂)。
1.2 材料与试剂:VP-ODS.6μm×250mm、Agela Technologies Inc.venusil XBP-C18μmA.6×250mm色谱柱;薄层层析硅胶GF254板(中国青岛海洋化工厂产预制板,规格cm×10cm批号、规格cm×20cm批号20060627);硅胶G板(青岛海洋化工厂分厂产预制板 , 规 格cm ×20cm,0.20μm ~0.25μm 批 号20050607);甲醇(色谱纯,Fisher公司),中性氧化铝(上海五四化学试剂有限公司),其它化学试剂均为分析纯。
1.3 供试品:尿通卡克乃其片(批号100601023、100411123、090323001、100115049、091104140、090815103、090509031、090607071、090923100编号依次为:NT1、NT2、NT3、NT4、NT5、NT6、NT7、NT8、NT9、NT10)共十批,由新疆A企业提供,甘草浸膏阴性对照(生产日期20100722)。
1.4 对照品:甘草酸铵对照品(批号:110731-200615、110731-200614供含量测定用),甘草次酸对照品(批号:110723-200310供含量测定用、批号:110723-200411供薄层扫描法含量测定用),甘草苷对照品(批号:111610-200604供含量测定用),均由中国药品生物制品检定研究院提供。
2 方法与结果
2.1 TLC鉴别方法研究
2.1.1 供试液的制备:供试品溶液:取尿通卡克乃其片1片,研细,甲醇mL,超声处理分钟(功率W,频率kHz),滤过,滤液蒸干,残渣加水mL使溶解,用乙醚振摇提取2次,每次25mL,弃去乙醚提取液,水层用水饱和正丁醇振摇提取2次,每次mL,合并正丁醇提取液,蒸干,残渣加甲醇1mL使溶解。
甘草浸膏阴性对照溶液:取甘草浸膏阴性样品1片量,研细,照供试品溶液同法制备,即得。对照品溶液:配制1mg·mL-1甘草酸铵对照品甲醇溶液。
2.1.2 TLC色谱条件:采用硅胶GF254板,乙酸乙酯-甲酸 -冰醋酸 -水(15:1:1:2)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视。
2.1.3 供试液的测定:照薄层色谱法(中国药典2010年版一部附录ⅥB)试验,吸取上述供试液各10μL,分别点于同一硅胶 GF254板上,照.1.2试验,结果见图,10批供试品色谱中,在与对照品色谱相应的位置上,均显相同的荧光熄灭斑点。甘草浸膏阴性对照样品无干扰。
2.1.4 不同点样浓度考察:如图2所示,供试品溶液点样量在10μL~30μL时的斑点较清晰,甘草浸膏阴性对照溶液在甘草酸铵对照品溶液相应位置无干扰。
2.1.5 检测限:取甘草酸铵对照品,加甲醇制成依次为0.16;0.32;0.48;0.64;0.80;1.0;1.5(mg·mL-1)的对照品溶液。分别于同一硅胶GF254薄层板上点样10μL,照
2.1.2 试验,结果上述浓度均可检出甘草酸铵斑点,但浓度在0.64 mg·mL-1以上的斑点较清晰。甘草酸铵薄层鉴别的检测限为(0.16~0.48)mg·mL-1,见图。
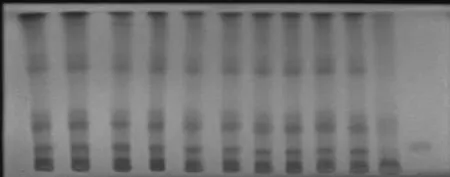
图1 10批尿通卡克乃其片中甘草酸铵TLC鉴别结果
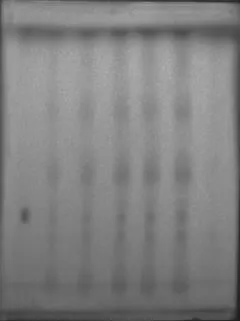
图2 尿通卡克乃其片中甘草酸铵TLC供试品点样浓度考察
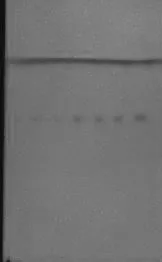
图3 尿通卡克乃其片中甘草酸铵TLC系统的检测限考察
2.2 HPLC鉴别方法研究
2.2.1 供试液的制备:供试品溶液:取尿通卡克乃其片1片,除去包衣,研细,置mL量瓶中,加流动相适量,超声处理10~30min(或振摇10min)使溶散,用流动相稀释至刻度,摇匀,滤过,滤液即得。
甘草浸膏阴性对照溶液:取甘草浸膏阴性样品1片量,研细,照供试品溶液同法制备,即得。对照品溶液的制备:另取甘草酸铵对照品,加流动相制成每1mL含0.14mg的溶液。
2.2.2 HPLC色谱条件:以十八烷基硅烷键合硅胶为填充剂,以甲醇 -0.2mol·L-1乙酸铵溶液 -冰乙酸(65:33:
1)为流动相(pH=5.0±0.5);柱温℃;检测波长为250nm。
2.2.3 供试液的测定:照高效液相色谱法(中国药典2010年版一部附录Ⅵ D)试验。分别精密量取2.2.1项下的供试液各20μL,照2.2.2项下方法试验,结果显示,供试品色谱中,在与甘草酸铵对照品溶液色谱相应的位置上,甘草浸膏阴性对照溶液无干扰,代表性图谱见图。

图4 尿通卡克乃其片-甘草浸膏HPLC色谱图
2.2.4 线性关系考察:精密称取甘草酸铵对照品适量用流动相溶解并稀释制成浓度系列,照2.2.2项下方法依法测定其峰面积(Y),以浓度为横坐标(X),进行回归,得线性方程,见表1。

表1 甘草酸铵对照品溶液线性方程及范围
2.2.5 检测限:直观法信噪比(S/N)3:1方法的最低检测限确定为.166μg,以信噪比(S/N)10:1确定最底检测量限为.416μg。见图。

图5 尿通卡克乃其片-甘草酸铵HPLC检测限
2.2.6 精密度试验:日内精密度(n=7)RSD为.6%;日间精密度(n=4)RSD为.4%。
2.2.7 稳定性试验:取同一供试品溶液分别于、1、2、6、12、26h,测定其峰面积,RSD为.8%(n=6),认为供试品溶液在26小时稳定。
2.2.8 耐用性试验:取同批供试品,比较振摇10min、超声min、10min、20min、30min的结果,除超声min时的结果差异较大外,其他处理条件样品间的结果RSD为1.6%~2.8%。
在柱温20℃~35℃条件下,改变流动相的比例、pH值、柱温、不同色谱柱(3根)、流速,在不同色谱仪(3台)上进行实验,均能重现,分离度均符合要求。经试验测得理论塔板数多在1200~3300之间,分离度均大于1.5,保留时间随甲醇比例的增减而提前或延后,一般在10min~20min之间。
2.2.9 回收率实验:精密称取尿通甘草浸膏阴性对照样品约0.5g(甘草酸铵理论含量为0)共9份,分别精密加入高、中、低甘草酸铵对照品溶液各3份,照.2.1项下同法制备供试品溶液,按2.2.2项下方法试验,结果:低(47.24μg·mL-1)、中(81.12μg·mL-1)、高(130.92μg·mL-1)3个浓度的添加回收率分别为.7%(RSD1.9%)、87.9%(RSD0.5%)、89.1%(RSD0.8%),3个浓度的平均回收率为85.9%,RSD=4.7%。
2.2.10 线性回收率实验:精密称取甘草酸铵对照品约12mg(11.81mg)加流动相溶解制成.4μg·mL-1的储备液(GD3)。另精密称取甘草浸膏阴性样品约0.5g份,分 别 精 密 加 入 GD3.0mL、1.0mL、2.0mL、3.0mL、5.0mL、10.0mL、13.0mL,照.2.1项下同法制备供试品溶液,按.2.2项下方法依法测定其峰面积(Y),以浓度为横坐标(X),进行回归,得线性指数方程,Y=141959e0.4591X (r=0.9804), 在.4~122.8μg·mL-1(0.188μg~2.456μg)范围内呈指数线性,见图。

图6 尿通卡克乃其片-甘草酸铵加样HPLC线性
3 讨论
3.1 鉴别用对照品或对照药材的选择 本制剂处方工艺中使用的是甘草浸膏,目前未生产甘草浸膏对照药材,其特征成分主要为甘草酸、甘草苷等,而其中甘草苷的含量较低,排除使用甘草苷对照品,而选择甘草酸铵、甘草次酸为对照品进行分析。为保持与《中国药典》一致,仅采用甘草酸铵对照品进行研究。同时建议开展甘草浸膏对照药材的研制工作。
3.2 TLC鉴别展开系统的选择 除2.1.2的TLC鉴别展开系统外,还采用硅胶GF254板,对文献报道[2-7]的种展开剂进行了研究。结果:展开系统[3]石油醚(60~90°C)-苯 -乙酸乙酯 -冰乙酸(10:22:6:0.45)适合甘草次酸成分的鉴别。
3.3 TLC鉴别供试品溶液制备方法的选择 除2.1.1的TLC鉴别供试品溶液制备方法外,还采用2.1.2的TLC鉴别展开系统,对文献报道[2-7]的7种供试品溶液制备方法进行了尝试。结果:方法[3]“取本品1片,研细,加盐酸mL及氯仿mL,超声min,滤过,蒸干,残渣加无水乙醇5mL溶解”适合甘草次酸的提取。
3.4 HPLC色谱系统的选择 除2.2.2所述的条件外,还考察了:①《中国药典2005年版二部》“复方甘草片”中甘草酸含量测定项下的色谱条件;文献[1]②中吗啡测定的色谱条件;③血竭测定的色谱条件。结果:均不适合甘草酸铵测定。
3.5 HPLC用供试品溶液制备方法的选择.2.1供试品溶液制备系采用《中国药典》2005年版一部“甘草浸膏”含量测定项下的供试品溶液制备方法,结合处方中甘草浸膏的用量,经换算将供试品的取用量改为1片量细粉,置25mL量瓶中,溶解后不再稀释,滤液直接测定。供试品溶液制备方法也曾采用水、甲醇、50%甲醇作为溶剂,但均不如采用流动相制备的供试品试液色谱图干扰少。
3.6 HPLC未能收载为含量测定方法的原因 在线性回收率实验中发现,在阴性对照取样量不变的条件下,加入甘草酸铵对照品的量与所测得的峰面积在一定范围内呈指数线性,其相关系数仅为0.9804,低于同系统单纯甘草酸铵对照品溶液线性相关系数0.9997,小于方法定量线性相关系数0.995。可能是造成低、中、高三种浓度的回收率差异较大的原因之一,制剂中的其他组分或成份对不同浓度甘草酸铵是否有吸附作用还有待于进一步研究,故未能收载为含量测定方法。
3.7 小结:HPLC与TLC比较,两者均采用甘草酸铵为对照品,色谱系统及色谱条件均与《中国药典2005年版一部》“甘草浸膏”相对应,但HPLC供试品的制备方法合理、对照品溶液浓度适宜,色谱柱类型、检测方式及参数合理,方法重现性较好,专属性、耐用性及灵敏度均有更强的优势,且可为将来指纹图谱研究奠定基础。
[1]卫生部药典委员会.药品标准-维吾尔药分册[S].1998.
[2]曾愠昀,彭中芳.紫雪颗粒甘草、玄参的薄层鉴别[J].中药与天然药物,2008,18(2)∶32-33.
[3]廖华卫,李瑞珍,陈飞苑.冷哮丸临床加减方颗粒中麻黄和甘草薄层色谱鉴别[J].现代食品与药品杂志,2006,16(4)∶11-12.
[4]万新,等.薄层色谱法同时鉴别龙胆泻肝汤制剂中甘草、当归、黄芩[J].华西药学杂志,1995,11(3):177.
[5]赖克道,等.薄层色谱法定性鉴别小柴胡颗粒中的柴胡、甘草[J].广西医学,2009,31(6):886-887.
[6]曾正宏,董明.复方硫酸软骨素片中甘草的薄层色谱鉴别[J].中国生化药物杂志,2002,23(5)∶251-253.
[7]韩家荣,等.复方甘草片质量控制探讨[J].中成药研究,1987,2:10-12.
猜你喜欢
中国交通信息化(2019年2期)2019-03-25 03:20:20
天然产物研究与开发(2019年1期)2019-03-01 05:41:26
中国交通信息化(2018年8期)2018-11-09 01:05:44
中国交通信息化(2018年6期)2018-08-29 01:19:34
中成药(2017年5期)2017-06-13 13:01:12
中国卫生标准管理(2015年4期)2016-01-14 05:16:45
西南医科大学学报(2016年4期)2016-01-03 01:26:29
中国交通信息化(2015年6期)2015-06-06 03:46:24
中国当代医药(2015年33期)2015-03-01 02:09:17
中国药业(2014年21期)2014-05-26 08:56:32
