
藻酸双酯钠对急性肺损伤模型小鼠的保护作用研究
2013-08-29 13:18史斌黄厚刚成都市第五人民医院麻醉科成都611130重庆医科大学附属永川医院麻醉科重庆402160
中国药房 2013年29期
史斌,黄厚刚(1.成都市第五人民医院麻醉科,成都611130;2.重庆医科大学附属永川医院麻醉科,重庆402160)
急性肺损伤/急性呼吸窘迫综合征(ALI/ARDS)是失控的全身炎症反应综合征(Systemic inflammatory response syndrome,SIRS)在肺部的具体表现[1]。近年研究[2-3]证明,在ALI/ARDS病程中原有的凝血与抗凝、纤溶与抗纤溶、炎症与抗炎之间的平衡被打破,凝血级联与炎症级联之间相互促进,形成一种自动放大的效应,使病情加重。因此干预凝血系统的异常对控制ALI/ARDS的病情发展具有重要意义。国内外有学者通过使用传统的抗凝药,如肝素治疗ALI/ARDS,取得了改善低氧血症和减轻炎症反应的效果[4-5]。类肝素类药物藻酸双酯钠(Polysaccharide sulphate,PSS)系海藻提取物,是一种新型类肝素海洋药物,其有明显的改善细胞变形、调节血脂代谢、抗凝等作用,同时有较强的分散乳化活性,且不易受外界因素的影响[6],临床上主要用于心脑血管疾病的治疗,但还未见用于预防ALI/ARDS的报道。本实验首先采用尾静脉注射脂多糖(LPS)建立小鼠ALI模型,然后尾静脉注射PSS,研究ALI/ARDS时PSS对小鼠炎症因子、信号转导分子的影响,以期为阐明PSS对ALI/ARDS的治疗作用及其分子机制提供参考。
1 材料
1.1 仪器
酶标仪(美国Bio-Rad公司);光学显微镜(日本Olympus公司);RM2016病理组织切片机(德国Leica公司);RC 5B高速低温离心机(美国Sorvall公司)。
1.2 药品与试剂
PSS原料药(中国青岛海尔药业公司,批号:20080405,纯度:99.9%);大肠埃希菌LPS(美国Sigma公司,批号:20060814S);肺组织中核转录因子-κB p65(NF-κB p65)免疫组化试剂盒(美国迈新公司);细胞间黏附因子1(ICAM-1)、肿瘤坏死因子α(TNF-α)酶联免疫吸附测定(ELISA)试剂盒(美国R&D公司)。
1.3 动物
健康C57BL/6小鼠40只,♂,体质量20~30g,购自华中科技大学同济医学院实验动物中心,合格证编号:SCXK(鄂)2008-0015。实验前禁食12h,自由饮水。
2 方法
2.1 分组与给药
将小鼠随机分为4组,每组10只。对照组,尾静脉注射0.9%氯化钠溶液(生理盐水);PSS组,根据文献[7]报道尾静脉注射PSS 10mg/kg;模型组,尾静脉注射LPS 10mg/kg;治疗组,先尾静脉注射LPS 10mg/kg后立即尾静脉注射PSS 10mg/kg。各组液体注射总量按10~20ml/kg给予。
2.2 检测指标
各组小鼠注射给药8h后,断颈法处死,立即打开胸腔,切取肺组织,取右肺上叶称质量,取左肺置于4%多聚甲醛溶液中固定,其余肺组织置入液氮中速冻后,-80℃冰箱保存。
2.2.1 肺组织湿干质量比值(W/D)。取各组小鼠右肺上叶肺组织用吸水纸吸干表面水分及血液后,称湿质量(W)后,置于94℃干燥箱内烘烤48h至恒质量,再称干质量(D),计算肺组织W/D。
2.2.2 肺组织形态学。取经固定后的每组小鼠的左肺组织,乙醇系列脱水,二甲苯透明,石蜡包埋,切片,厚度3μm,苏木精-伊红染色,中性树胶封片,在光学显微镜下观察肺组织形态学变化。
2.2.3 肺组织中TNF-α、ICAM-1的浓度。取各组小鼠冰冻肺组织100mg,充分匀浆,离心后取上清液,采用ELISA法测定肺组织中TNF-α和ICAM-1的浓度,具体按试剂盒要求操作。
2.2.4 肺组织中NF-κB p65活性。采用免疫组化法,按试剂盒说明书操作,加入兔抗NF-κB p65抗体(1∶200),阴性对照采用正常山羊血清代替,以光密度(OD)值为指标检测各组小鼠肺组织中NF-κB p65活性。采用图像分析法,通过显微镜放大10×10倍摄取图像,观察细胞着色情况,着色越明显,NF-κB p65表达强度越高。
2.3 统计学处理
3 结果
3.1 肺组织W/D变化
与对照组比较,模型组、治疗组小鼠肺组织的W/D均明显增加(P<0.05);PSS组小鼠肺组织的W/D差异无统计学意义(P>0.05)。与模型组比较,治疗组小鼠肺组织的W/D明显减小(P<0.05),具体结果详见表1。
3.2 肺组织形态学变化
表1 各组小鼠肺组织W/D及ICAM-1、TNF-α、NF-κB p65水平比较(±s,n=10)Tab 1Comparison of W/D and ICAM-1,TNF-α and NF-κB p65levels in lung tissue of mice in each group(±s,n=10)

表1 各组小鼠肺组织W/D及ICAM-1、TNF-α、NF-κB p65水平比较(±s,n=10)Tab 1Comparison of W/D and ICAM-1,TNF-α and NF-κB p65levels in lung tissue of mice in each group(±s,n=10)
与对照组比较:*P<0.05,**P<0.01;与模型组比较:#P<0.05,##P<0.01vs.control group:*P<0.05,**P<0.01;vs.model group:#P<0.05,##P<0.01
组别对照组PSS组模型组治疗组W/D 4.13±0.234.21±0.245.35±0.37*5.05±0.33*#ICAM-1,ng/L 59.4±7.461.6±7.1185.5±18.8*75.5±14.5*#TNF-α,ng/L 258.5±15.7278.4±23.3522.3±35.4*405.2±26.6*#NF-κB p65,ng/L 187.25±21.41190.58±19.34256.42±24.45**225.91±20.12*##
对照组和PSS组小鼠肺组织结构完整,肺泡间隔适中,无水肿、炎症,肺泡腔清晰;模型组小鼠肺泡间隔增宽,肺泡壁破坏,肺间质水肿,部分肺泡融合,腔内可见渗出、水肿及出血,肺间质炎细胞浸润;治疗组小鼠肺组织上述病理变化较模型组明显改善,肺泡壁破坏减少,出血及渗出减轻。显微镜图见图1。
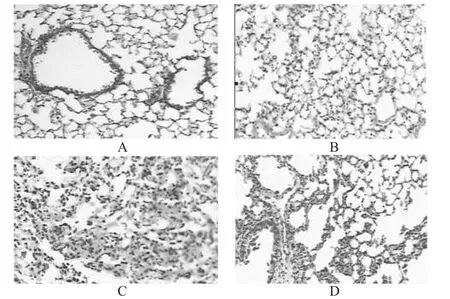
图1 各组小鼠肺组织病理学变化(HE,×100)A.对照组;B.PSS组;C.模型组;D.治疗组Fig 1 Pathological change of lung tissue of mice in each grou(pHE,×100)A.control group;B.PSS group;C.model group;D.treatment group
3.3 肺组织中ICAM-1、TNF-α的浓度变化
与对照组比较,模型组、治疗组小鼠肺组织中ICAM-1、TNF-α浓度均明显增加(P<0.05),PPS组小鼠肺组织中上述指标的差异无统计学意义(P>0.05)。与模型组比较,治疗组小鼠肺组织中ICAM-1、TNF-α浓度均明显减小(P<0.05),具体结果详见表1。
3.4 肺组织中NF-κB p65活性变化
镜下观察,对照组和PSS组小鼠肺组织中NF-κB p65表达主要集中在肺泡和巨噬细胞等中,呈浅黄色,以胞浆为主;模型组小鼠肺组织中NF-κB p65表达主要集中在炎症细胞和支气管上皮细胞等中,胞浆和胞核的着色明显加深,呈棕黄色,且胞核染色明显增多,表达的强度与对照组比较差异有统计学意义(P<0.01)。与模型组比较,治疗组小鼠肺组织中NF-κB p65表达明显降低(P<0.01),结果见表1,显微镜图见图2。
4 讨论

图2 各组小鼠肺组织中NF-κB p65的染色情况(×400)A.对照组;B.PSS组;C.模型组;D.治疗组Fig 2 Staining of NF-κB p65in lung tissue of mice in each grou(p×400)A.control group;B.PSS group;C.model group;D.treatment group
内毒素是ALI的主要致病因素之一,是革兰阴性菌细胞外膜成分,其主要致病物质LPS,具有严重的致炎作用。目前认为在ALI/ARDS早期,血管内皮细胞以及单核巨噬细胞在LPS的诱导下可表达TNF-α等细胞因子以及ICAM-1等多种黏附分子。TNF-α和ICAM-1介导多形核白细胞(PMN)与肺微血管内皮细胞黏附后,既可激活PMN释放弹性蛋白酶等炎性介质降解肺微血管内皮细胞(PMVEC)结构蛋白[8],又能启动ICAM-1信号转导重构PMVEC细胞骨架[9],致使PMVEC正常的细胞结构和功能受到破坏,通透性增加,形成渗透性肺水肿,导致肺顺应性下降和氧合障碍,引发内毒素性ALI。
NF-κB在细胞质中与其抑制蛋白IκB结合以非活性异二聚体形式存在,受细胞因子TNF-α或细菌、病毒、缺氧等刺激后,导致IκB磷酸化而降解。IκB降解使NF-κB游离,转位到核内,与靶基因上的κB位点结合而启动基因转录,发挥一系列细胞效应[10]。本研究在给予LPS后8h,免疫组化定位显示NF-κB活化转位至细胞核内,上调TNF-α和ICAM-1浓度,这与文献[11]结果一致。给予治疗剂量的PPS后,抑制了NF-κB p65的活化,说明PPS对ALI模型小鼠的保护作用通过抑制NF-κB p65细胞信号通路发挥效应。
与对照组比较,PSS组小鼠未见明显的肺损伤以及TNF-α、ICAM-1和NF-κB的相应改变,说明在实验剂量(10mg/kg)内对小鼠无明显副作用。
综上所述,PPS能对LPS诱导的ALI模型小鼠具有保护作用,可能是通过抑制NF-κB p65的活化、下调TNF-α和ICAM-1浓度而发挥作用。但相关的具体作用机制以及其保护作用与剂量的相关性有待进一步的研究。
[1] Jackson JC,Hopkins RO,Miller RR,et al.Acute respiratory distress syndrome,sepsis,and cognitive decline:a review and case study[J].South Med J,2009,102(11):1150.
[2] Chambers RC.Procoagulant signalling mechanisms in lung inflammation and fibrosis:novel opportunities for pharmacological intervention?[J].Br J Pharmacol,2008,153(Suppl 1):S367.
[3] MacLaren R,Stringer KA.Emerging role of anticoagulants and fibrinolytics in the treatment of acute respiratory distress syndrome[J].Pharmacotherapy,2007,27(6):860.
[4] Wang M,He J,Mei B,et al.Therapeutic effects and antiinflammatory mechanisms of heparin on acute lung injury in rabbits[J].Acad Emerg Med,2008,15(7):656.
[5] 王美堂,何建,梅冰,等.肝素对急性肺损伤/急性呼吸窘迫综合征的治疗作用[J].中国急救医学,2007,27(5):443.
[6] 赵仁亮,王春霞,赵俊武,等.藻酸双酯钠对血管内皮细胞的生长及其与中性粒细胞黏附功能的影响[J].中国海洋药物杂志,2007,26(3):41.
[7] 游咏,谢红炬,张岳峰,等.大鼠脑缺血-再灌注损伤中P-选择素的表达对脑血流的影响[J].中风与神经疾病杂志,2005,22(2):155.
[8] Gonzalez RJ,Moore EE,Ciesla DJ,et al.Post-hemorrhagic shock mesenteric lymph activates human pulmonary microvascular endothelium for in vitro neutrophil-mediated injury:the role of intercellular adhesion molecule-1[J].J Trauma,2003,54(2):219.
[9]Wang Q,Yerukhimovich M,Gaarde WA,et al.MKK3and-6-dependent activation of p38alpha MAP kinase is required for cytoskeletal changes in pulmonary microvascular endothelial cells induced by ICAM-1ligation[J].Am J Physiol Lung Cell Mol Physiol,2005,288(2):359.
[10] Jijon H,Allard B,Jobin C.NF-kappaB inducing kinase activates NF-kappaB transcriptional activity independently of IkappaB kinase gamma through a p38MAPK-dependent RelA phosphorylation pathway[J].Cell Signal,2004,16(9):1023.
[11] 余跃,杜烨玮,贺晶,等.MAPK和NF-κB上调脂多糖诱导的小鼠肺微血管内皮细胞Toll-like受体4的表达[J].基础医学与临床,2006,26(12):1328.
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
昆明医科大学学报(2022年1期)2022-02-28
昆明医科大学学报(2021年10期)2021-12-02
现代临床医学(2021年5期)2021-11-02
天津医科大学学报(2021年4期)2021-08-21
现代临床医学(2021年4期)2021-07-31
科学大众(2021年6期)2021-07-20
学苑创造·A版(2020年9期)2020-10-13
中国眼镜科技杂志(2019年9期)2019-11-11
娃娃乐园·综合智能(2019年3期)2019-04-03
