
氮化硅胶的制备及其吸收CO2性能的研究
2013-08-14 09:08原艳芬王树国
化学与生物工程 2013年2期
原艳芬,王树国,李 琳,伍 明
(中南民族大学化学与材料科学学院,湖北 武汉430074)
近年来,由于煤炭、石油、天然气等含碳物质的燃烧、氧化等释放出大量的CO2,使得大气中的CO2含量逐年上升。据统计,大气中CO2浓度以每年0.15%的速度递增,温室效应日益严重[1-3]。
目前,分离回收CO2的方法有吸收法、吸附法、膜分离法等[4,5]。化学吸收法因其吸收效率高、处理量大而得到广泛深入的研究和应用。其中有机胺工艺始于20世纪30年代,以胺类化合物吸收CO2,具有吸收量大、吸收效果好、洗涤剂可循环利用并能回收到高纯产品的特点,但吸收剂在运行中损失大、易氧化损耗、腐蚀设备、易发泡、成本偏高。因此,研究者考虑使用固体吸收剂,如将氨丙基或其它仲胺基、叔胺基接枝到硅胶[6]以及氧化硅介孔分子筛如 HMS、SBA-15、SBA-12、SBA-16、MCM-41 等 材 料 上[7-11],利 用 氨 基(或胺基)与CO2反应来吸收CO2,既具有有机胺的功能,能在较低温度下吸收和解吸,又克服了液体吸收剂的不足。但这类吸收剂的性能与接枝的氨(胺)基数量有关,由于硅基材料表面硅羟基数量的限制,接枝的基团量较低;同时,接枝的试剂有毒、价格昂贵、接枝条件苛刻,当温度超过200℃时,有机官能团容易分解,使其工业应用受到限制。
在高温下利用氨气使硅氧化物氮化,可在其表面形成-NHx基团;由于Si-O-Si可断开而连接-NHx基团,因此可以形成更多的氨基。Lednor等利用ZSM-5、商业硅胶分别与氨气在1100℃反应4h,得到ZSM-5的氮含量(质量分数)为4%、硅胶的氮含量为22%;将这些氮化产物作为固体碱催化剂催化Knoevenagel反应,表现出良好的催化性能,并且随着氮含量的增加而提高。Wang等[12]用这种方法使SBA-15介孔分子筛氮化,氮含量达到20%,该氮化物在600℃仍保持稳定。Zhao等[13]在SBA-15氮化后,利用氨基的作用将银负载到分子筛孔道中。
作者在此采用高温通氨气的方法使氧化硅氮化,并将氮化的氧化硅用于吸收CO2,优化了氮化硅胶的合成条件,并对氮化硅胶吸收CO2的性能进行了测试,以期为氮化硅胶的工业应用提供理论基础。
1 实验
1.1 试剂与仪器
正硅酸四乙酯(TEOS,分析纯)、无水乙醇(分析纯)、氨水(NH325%~28%,分析纯)、聚乙二醇(PEG200,化学纯),上海国药集团;高纯氨气(纯度99.999%),大连安瑞森特种气体化学品有限公司。
Nicolet NEXUS-670型傅立叶红外光谱仪;Hitachi S-4800 型场发射扫描 电 子 显 微镜;VG Multilab 2000型X-射线光电子能谱仪,美国Thermal Electron公司;AV 400型核磁共振波谱仪,瑞士Bruker公司;Vario Micro cube型元素分析仪,德国Elementar公司。
1.2 氮化硅胶的制备
1.2.1 硅胶的制备
以TEOS、无水乙醇和蒸馏水为原料,以氨水为催化剂,按一定体积比(TEOS∶无水乙醇∶水∶氨水=6mL∶40mL∶20mL∶12mL)混合,以PEG200(添加量为TEOS质量的10%)为改性剂,在333K下充分搅拌,混合均匀,然后将混合溶液放入密闭容器内,置于恒温(333K)水浴中使其凝胶化。将湿凝胶先在393K下干燥3h,再在873K下处理4h得到干凝胶,标记为SiO2-40。
将无水乙醇与水的比例改为50mL∶10mL、60 mL∶0mL,所制得的干凝胶分别标记为SiO2-50、SiO2-60。
1.2.2 硅胶的氮化
将硅胶SiO2-60置于氧化铝瓷舟中,然后置于氧化铝瓷管中,再将其放在以硅碳管加热的管式炉中。通入N2,抽真空3次,以5℃·min-1的升温速率程序升温至400℃;切换为 NH3(流速为100mL·min-1),在目标温度分别为1000℃、900℃、800℃、700℃下氮化20h,再在N2环境中冷却至室温,即制得氮化硅胶,分别标记为 SiO2-N-1000、SiO2-N-900、SiO2-N-800 、SiO2-N-700。
1.3 吸收CO2性能测试
将制得的氮化硅胶装至吸收柱内,在N2氛围中200℃下预处理0.5h(N2流量设为30mL·min-1);冷却至吸收温度30℃;温度达到预定值时,进样气体由N2转换成CO2/N2(CO215%,体积分数)混合气体,吸收1h;吸收完毕后,进样气改为N2,200℃下将已吸收饱和的吸收剂进行解吸,利用气相色谱仪检测解吸的气体量,以此来表征氮化硅胶对CO2的吸收能力。
2 结果与讨论
2.1 硅胶的表征
在不同无水乙醇和水配比下所制得的硅胶的SEM照片及其粒径分布见图1。
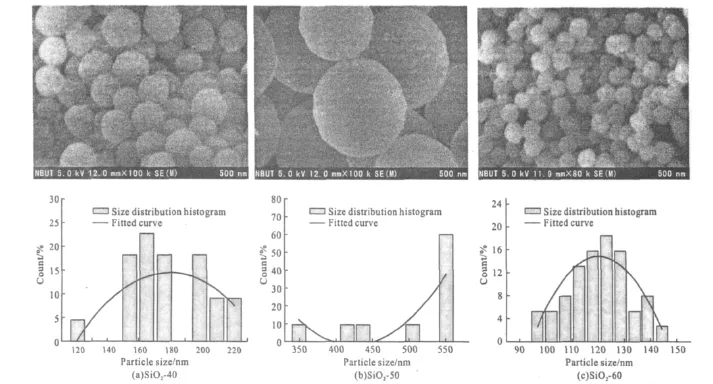
图1 硅胶的SEM照片及其粒径分布图Fig.1 SEM Images and the particle size distribution of the silica gels
由图1可知,不同无水乙醇和水配比下制得的硅胶均为实心球状,但是配比不同,所制得的球状硅胶的平均粒径不同、粒径分布也不同,在无水乙醇与水的配比分别为40mL∶20mL(图1a)、50mL∶10mL(图1b)、60mL∶0mL(图1c)时,所得到的硅胶粒径分别为179.87nm、499.61nm、120.35nm。说明合成时水量的不同直接导致了球形氧化硅粒径大小的差异。在无水乙醇与水配比为60mL∶0mL时,所得到的硅胶粒径最小、比表面积较大、粒径分布更集中,更利于后续的氮化实验。
2.2 氮化硅胶的表征
2.2.1 元素分析
在氮化时间为20h,氮化温度分别为1000℃、900℃、800℃、700℃时所制备的氮化硅胶的氮含量分别为9.48%、7.04%、1.48%、0.87%。由此可知,随着氮化温度的升高,氮化硅胶的氮含量逐渐增加。通入氨气对氧化硅进行氮化处理,是利用氮原子部分取代氧原子从而得到氮氧化硅材料的,由于氮原子相对于氧原子有更低的电负性,使得氮氧化硅具有了Lewis碱性。由元素分析结果可知,取代氧化硅中的酸性位需要在较高温度下保持一定时间才可完成。
2.2.229Si MAS NMR谱图分析(图2)
由图2可看出,氮化硅胶样品在-54ppm、-72 ppm和-89ppm处出现3个峰,分别归属于SiN3O、SiN2O2、SiNO3[14]。氮化硅胶样品中Si的主要化学环境为SiN3O,而氮化前硅胶在-109ppm、-100ppm处归属于SiO4和SiO3(OH)的2个特征峰基本消失[12]。表明氮化过程中电负性较低的氮原子部分取代了硅胶中的氧原子,从而进入到了硅胶骨架中[15]。
2.2.3 XPS谱图分析(图3)
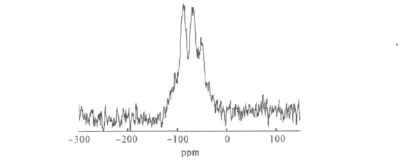
图2 氮化硅胶SiO2-N-1000的29Si MAS NMR图谱Fig.2 29Si MAS NMR Spectrum of the nitrided silica gel SiO2-N-1000
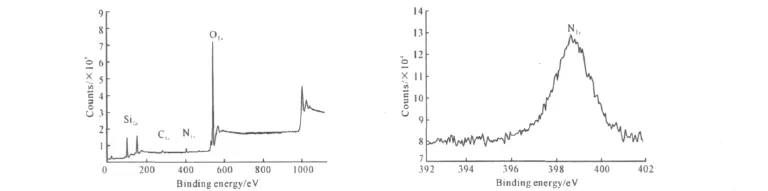
图3 氮化硅胶SiO2-N-1000的XPS图谱Fig.3 XPS Patterns of the nitrided silica gel SiO2-N-1000
由图3可看出,氮化硅胶样品在103.7eV、398.7eV和533.0eV处的主峰分别归属为Si2p、N1s和O1s峰,与此同时,也可在285eV处观察到C1s峰。标准图谱中N1s峰通常位于397.9eV处,而本实验位于398.7 eV,表明部分氮原子以-NH2形态存在;同时,在氮化硅胶中氮原子还可能以=N-或-NH-形态存在,具体存在形态尚不能确定[16]。
2.2.4 红外光谱分析
硅胶SiO2-60氮化前后的红外光谱见图4。
由图4SiO2-60的红外光谱可看出,3747.66cm-1附近的吸收带归属于Si-OH的伸缩振动,3624.26 cm-1附近的吸收带归属于氢键羟基[17,18],1115.31 cm-1附近的吸收带归属于Si-O-Si的非对称伸缩振动,809.32cm-1处的吸收带归属于三维结构中对称Si-O-Si的弯曲振动,1628.96cm-1附近的吸收带归属于HOH吸附水的弯曲振动,3446.34cm-1附近的吸收带归属于水的羟基的伸缩振动。由SiO2-N-1000的红外光谱可看出,位于Si-OH的特征吸收峰基本消失,而在3378.73cm-1附近出现了新的吸收峰,归属于≡Si-NH-Si≡硅氮烷中-NHx的伸缩振动。表明,硅胶氮化后几乎所有的表面硅羟基均与氨气反应,形成了-NHx基团[13]。
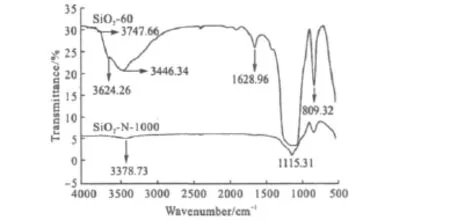
图4 硅胶SiO2-60氮化前后的红外光谱Fig.4 FTIR Spectra of SiO4-60before and after nitridation
2.3 对CO2的吸收性能
在不同吸收温度下,硅胶氮化前后对CO2的吸收能力见图5。
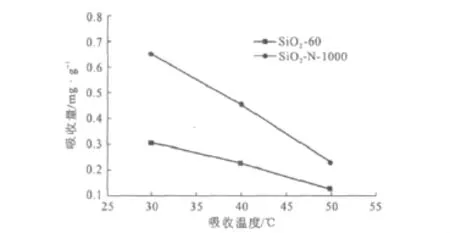
图5 不同吸收温度下,硅胶氮化前后对CO2的吸收能力Fig.5 Absorption capacity of silica gel before and after nitridation at different absorption temperatures
由图5可看出,氮化后的硅胶对CO2的吸收能力明显强于未氮化的硅胶,这是由于氮化硅胶中碱性-NHx基团和酸性气体CO2之间的强吸收所致;随着吸收温度的不断升高,吸收能力不断下降。这是由于吸收过程为放热反应,升高温度不利于吸收的进行,使得吸收剂的吸收能力下降,在温度为30℃时,吸收量最大,为6.71×10-4g·g-1,远高于未氮化硅胶的3.08×10-4g·g-1。
氮化硅胶SiO2-N-1000在30℃下吸收CO2的重现性实验结果见图6。
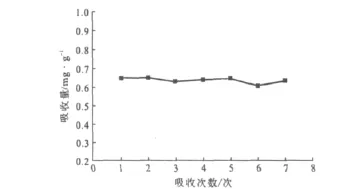
图6 SiO2-N-1000吸收CO2的重现性实验结果Fig.6 Reproducible experiment result for CO2absorption by SiO2-N-1000
由图6可看出,氮化硅胶SiO2-N-1000经过连续7次吸收-解吸后,对CO2的吸收能力未有明显的降低,说明氮化硅胶SiO2-N-1000吸收CO2的重复利用性较高。
3 结论
通过高温下通入氨气对硅胶进行氮化改性,得到了含氮量高的氮化硅胶。在氨气流速为100mL·min-1、氮化温度为1000℃、氮化时间为20h的条件下,氮化硅胶的氮含量最高达9.48%。氮化硅胶SiO2-N-1000对CO2的吸收量达6.71×10-4g·g-1,较未氮化硅胶(3.08×10-4g·g-1)明显提高,且重复利用性较高。
[1]Lashof D A,Ahuja D R.Relative contributions of greenhouse gas emissions to global warming[J].Nature,1990,344(6266):529-531.
[2]Ehlers E,Krafft T.Understanding the Earth System[M].Berlin:Springer-Verlag,2001:17-55.
[3]Song C S.Global challenges and strategies for control,conversion and utilization of CO2for sustainable development involving energy,catalysis,adsorption and chemical processing[J].Catalysis Today,2006,115(1-4):2-32.
[4]Garcia-Ricard O J,Hernandez-Maldonado A J.Cu2(Pyrazine-2,3-dicarboxylate)2(4,4′-bipyridine)porous coordination sorbents:Activation temperature,textural properties,and CO2adsorption at low pressure range[J].J Physical Chemistry C,2010,114(4):1827-1834.
[5]Meis N N A H,Bitter J H,de Jong K P.Support and size effects of activated hydrotalcites for precombustion CO2capture[J].Ind Eng Chem Res,2010,49(3):1229-1235.
[6]Leal O,Bolivar C,Ovalles C,et al.Reversible adsorption of carbon dioxide on amine surface-bonded silica gel[J].Inorganica Chimica Acta,1995,240(1-2):183-189.
[7]Knowles G P,Graham J V,Delaney S W,et al.Aminopropyl-functionalized mesoporous silicas as CO2adsorbents[J].Fuel Processing Technology,2005,86(14-15):1435-1448.
[8]Hiyoshi N,Yogo K,Yashima T.Adsorption characteristics of carbon dioxide on organically functionalized SBA-15[J].Microporous and Mesoporous Materials,2005,84(1-3):357-365.
[9]Zelenak V,Halamova D,Gaberova L,et al.Amine-modified SBA-12mesoporous silica for carbon dioxide capture:Effect of amine basicity on sorption properties[J].Microporous and Mesoporous Materials,2008,116(1-3):358-364.
[10]Knofel C,Descarpentries J,Benzaouia A,et al.Functionalised micro-/mesoporous silica for the adsorption of carbon dioxide[J].Microporous and Mesoporous Materials,2007,99(1-2):79-85.
[11]He Y,Seaton N A.Heats of adsorption and adsorption heterogeneity for methane,ethane,and carbon dioxide in MCM-41[J].Langmuir,2006,22(3):1150-1155.
[12]Wang Jiacheng,Liu Qian.Structural change and characterization in nitrogen-incorporated SBA-15oxynitride mesoporous materials via different thermal history[J].Microporous and Mesoporous Materials,2005,83(1-3):225-232.
[13]Zhao Yinfeng,Qi Yue,Wei Yingxu,et al.Incorporation of Ag nanostructures into channels of nitrided mesoporous silica[J].Microporous and Mesoporous Materials,2008,111(1-3):300-306.
[14]Haskouri J E,Cabrera S,Sapina F,et al.Ordered mesoporous silicon oxynitrides[J].Advanced Materials,2001,13(3):192-195.
[15]Bendjeriou-Sedjerari A,Pelletier J D A,Abou-hamad E,et al.A well-defined mesoporous amine silica surface via a selective treatment of SBA-15with ammonia[J].Chemical Communications,2012,48(25):3067-3069.
[16]Benitez J J,Daz A,Laurent Y,et al.Characterisation,surface hydrolysis and nitrogen stability in aluminophosphate oxynitride(AlPON)catalysts[J].Applied Catalysis A:General,1999,176(2):177-187.
[17]Tripp C P,Hair M L.Direct observation of the surface bonds be-tween self-assembled monolayers of octadecyltrichlorosilane and silica surfaces:A low-frequency IR study at the solid/liquid interface[J].Langmuir,1995,11(4):1215-1219.
[18]Fripiat N,Centeno M A,Grange P.Identification and stability of the nitrogenous species in zirconium phosphate oxynitride catalysts[J].Chemistry of Materials,1999,11(6):1434-1445.
猜你喜欢
陶瓷学报(2021年5期)2021-11-22
陶瓷学报(2021年4期)2021-10-14
陶瓷学报(2019年6期)2019-10-27
统计与决策(2017年2期)2017-03-20
无机盐工业(2017年4期)2017-03-10
电子制作(2017年24期)2017-02-02
癌变·畸变·突变(2016年4期)2016-08-22
材料科学与工程学报(2016年5期)2016-02-27
浙江理工大学学报(自然科学版)(2015年2期)2015-03-01
湘潭大学学报(哲学社会科学版)(2014年6期)2014-02-28
