
玉米MIR基因转录起始位点预测方法研究
2013-05-10 02:07张芳芳王洪秋付志远李卫华
河南农业大学学报 2013年1期
张芳芳,王洪秋,付志远,李卫华,丁 冬
(河南农业大学农学院,河南郑州450002)
miRNA是由生物自身基因组序列中的miRNA基因(MIR基因)编码的,长度为19~23 nt(nucleotide)的non-coding RNA.近年来,随着miRNA作为一种关键的转录和转录后水平的调节因子的作用被阐明,miRNA越来越受到科研工作者的关注[1].miRNA的作用方式,是通过识别并降解与其自身序列互补的mRNA,调控在特定环境条件下、特定发育时期,特定组织或器官中miRNA靶基因的丰度,并最终影响性状的表现.植物中已报道了多于1 000 个 miRNA(http://microrna.sanger.ac.uk/Release 18.0)[2].植物 miRNA 处于基因表达网络的上游,作为一种反式作用因子,通过控制关键调节蛋白质的含量来影响个体的发育特性,如拟南芥中已知的miRNA在花器官中都有表达,如miR 393在所有组织中都能检测到,而其在花器官中的表达量最大[3].miRNA的基因表达调控作用同样也被证实在植物激素的响应中起到关键作用,如水稻中的ARF 8对生长素水平的负反馈调节是通过调节GH3基因实现的,而 miR 167介导的 ARF 8和ARF 17转录本剪切可控制GH3家族蛋白质的含量,从而控制自由态生长素的水平,进而影响植物的发育[4].MIR基因被证实是由RNA聚合酶Ⅱ指导转录的,在这类基因核心启动子区域具有保守的顺式作用元件如 CAAT-box 和 TATA-box[5],XIE等[6]通过检测52个拟南芥MIR基因发现,在大多数拟南芥MIR基因核心启动子区域具有TATA-box.ZHOU 等[7]系统分析了人类、线虫、拟南芥和水稻4种模式生物MIR基因的核心启动子区域,证实在动物MIR基因启动子中,TATA-box并非保守,而在植物中,MIR基因启动子区域的TATA-box是非常保守的.在分子生物学研究过程中,要明确MIR基因的表达调控机制,首先要确定其启动子区域,而启动子区域的确定有赖于转录起始位点的确定.在模式植物拟南芥中,转录起始位点全部位于pre-miR茎环结构上游3 kb以内,多数集中于目标基因上游的0.5~2 kb序列[7].由于玉米的基因组测序工作完成时间较短,目前尚无关于玉米MIR基因表达调控的研究报道,而且,由于非编码基因与蛋白质编码基因的不同,尚未见到关于MIR基因转录起始位点的系统报道.本研究中开发出一种转录起始位点的预测方法,并使用这种方法对玉米胚萌发阶段杂种优势相关miRNA进行了分析.
1 材料与方法
1.1 玉米MIR基因信息分析
本研究所利用的玉米全基因组序列为亚利桑那大学公布的自交系B 73基因组Pseudomoleculars(http://www2.genome.arizona.edu/genomes/maize,序列版本号1.0).玉米成熟 miR及pre-miR的茎环结构从miRBase网站(http://www.mirbase.org/)获得,并在 maizeGDB网站(http://www.maizegdb.org/)使用Blast工具进行在线染色体定位(参考序列为玉米基因组Version1.0),得到定位的pre-miR茎环序列在玉米基因组序列中找到相对应的区域,获得pre-miR茎环结构正向序列上游5 000 bp的序列,该5 000 bp序列作为转录起始位点预测的起始序列.
1.2 基本启动子区域保守顺式位点分析
利用 plantCARE(http://bioinformatics.psb.ugent.be/webtools/plantcare/html/)[8]在线分析工具,对pre-miR茎环结构上游5 000 bp序列分段进行顺式作用元件分析.在获得的顺式元件中寻找TATA-box和CAAT-box,设定这两者之间的距离为30~60 nt的为可能的顺式作用位点,则TATA-box下游30 nt处为转录起始位点.
1.3 5’-RACE(快速扩增cDNA末端)证实这种预测方法的准确性
玉米自交系B 73植株在实验室培育至二叶一心期,取其幼嫩的地上部分在液氮中研磨至细碎,使用TRIzol试剂(Invitrogen,USA)提取其组织总RNA.5’RACE使用大连宝生物公司生产的5’-Full RACE试剂盒进行(Takara,Jap).操作方法详见产品说明书.进行扩增的基因特异引物设计在茎环结构内部,5’端引物由试剂盒提供.
2 结果与分析
2.1 玉米MIR基因在染色体上的分布
目前报道的玉米miRNA共有为172个.在前期的研究中,利用Solexa深度测序技术,分析了玉米胚萌发过程中杂种优势相关miRNA的表达谱,在萌发阶段的玉米杂交种和自交系中同时检测到的107个保守miRNA.将这107个miRNA的茎环结构在玉米基因组中进行了定位分析,发现这些MIR基因分布于全部10条染色体中(图1和表1).

图1 MIR基因在玉米染色体上的分布情况Fig.1 Maize MIR genes location in each chromosome
2.2 玉米已知MIR转录起始位点的预测结果
用特征Cis位点分析的方法,对107个玉米MIR基因的启动子区域进行了顺式作用位点分析,并根据这些顺式位点的分布规律,提出MIR基因的转录起始位点预测方法,即MIR基因基本启动子区域具有典型的CAAT-box和TATA-box结构,而且,这2种结构分别位于转录起始位点上游75和30 nt处.利用上述预测方法,对107个玉米胚萌发阶段杂种优势相关miRNA的编码MIR基因进位点.另外22个MIR基因由于已报道的测序玉米自交系B 73中序列不完整(在其茎环结构上游具有未测通的碱基序列)而未能预测其转录起始位点.对这85个MIR基因的转录起始位点分析发现,这些基因的转录起始位点距茎环结构都在5 kb之内,大多(80%)分布在2 000 nt以内(图2),这与拟南芥中的MIR基因转录起始位点的分布规律相同.在玉米中,MIR基因同样倾向于使用嘌呤碱基(A和G)起始基因的转录(图3),这与其他由RNA聚合酶Ⅱ指导转录的RNA(如mRNA)相同.

表1 保守顺式元件及转录起始位点与pre-miR茎环间的距离Table 1 Distance between transcription starting site(TSS),conserved cis elements and pre-miR
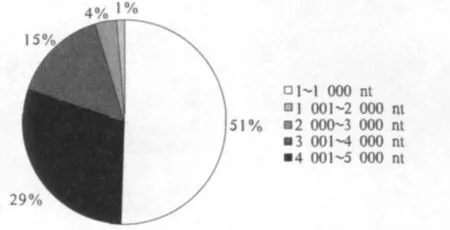
图2 MIR基因转录起始位点与pre-miR茎环结构5’端的距离Fig.2 Distance between MIR gene transcription starting site and pre-miR stem-loop 5’ends

图3 MIR基因转录起始位点的碱基使用情况Fig.3 MIR gene transcription starting site base bias
2.3 5’RACE扩增证实玉米MIR基因转录起始位点预测结果
用5’RACE方法,对其中5个MIR基因(分别为 MIR156k,159a,160b,164a,167c)转录起始位点进行了验证.发现用本预测方法获得的结果与试验结果相吻合(图4).在序列5’端用颜色标明的碱基为用5’RACE方法扩增并测序得到的转录本转录起始位点.在5个例子中,试验获得的5’端与用预测方法得到的转录起始位点相一致.

图4 5’RACE获得的5个MIR转录本5’端序列Fig.4 Five 5’end sequences of MIR transcripts obtained via 5’RACE
3 结论与讨论
迄今为止,功能得到明确证实的玉米miRNA还很少,在玉米中得到功能确证的miRNA主要有:miR 166和 miR 390调控茎的发育[9],miR172控制花器官的发育[10,11].此外,一些玉米 miRNA 也被证实受胁迫条件诱导,参与一些非生物胁迫的细胞响应,并在转录水平调节非生物胁迫响应基因的表达[12~15].miRNA 的调节作用据信还参与玉米杂种优势的形成,如miR167在2个自交系的植株和子粒中具有相似的表达水平,但与F1的表达模式相差很大[16].
mRNA降解的一种保守机制即通过miRNA靶定特定的转录本并对其进行降解.miR调节的基因表达常呈现家族共同调节的态势,暗示miRNA的表达也许是存在共同的调节因子的.共同的调节因子可能是激素或转录因子,它们共同的特征是识别启动子区域的特异cis位点,因此,确定其启动子区域对研究MIR基因的表达调控具有重要意义.
mRNA的预测已有成熟的方法,如 Fgenesh(http://linux1.softberry.com/)等,而针对特定类型的非编码基因的转录预测尚未出现.本研究依据MIR基因启动子保守cis位点提出的玉米MIR基因转录起始位点的预测方法,并通过试验手段证实了该方法的可靠性,为进一步实现MIR基因表达调控的研究提供了基础.开发出的这种MIR基因转录起始位点预测方法将有助于玉米microRNA的表达调控研究.
[1] WATERHOUSE P M,FUSARO A F.Plant science.Viruses face a double defense by plant small RNAs[J].Science,2006,313:54 -55.
[2] GRIFFITHS-JONES S,SAINI H K,VAN DONGEN S,et al.miRBase:tools for microRNA genomics [J].Nucleic Acids Res,2008,36:154 -158.
[3] SUNKAR R,ZHU J K.Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis[J].Plant Cell,2004,16(8):2001 -2019.
[4] YANG J H,HAN S J,YOON E K,et al.Evidence of an auxin signal pathway,microRNA167-ARF8-GH3,and its response to exogenous auxin in cultured rice cells[J].Nucleic Acids Res,2006,34(6):1892 -1899.
[5] JONE-RHOADES M W,BARTEL D P.Computational identification of plant microRNAs and their targets,including a stress-induced miRNA [J].Mol Cell,2004,14(6):787-99.
[6] XIE Z,ALLEN E,FAHLGREN N,et al.Expression of Arabidopsis MIRNA genes[J].Plant Physiology,2005,138(4):2145-2154.
[7] ZHOU X,RUAN J,WANG G,et al.Characte-rization and identification of microRNA core promoters in four model species[J].PLoS Comput Biol,2007,3(3):e37.
[8] LESCOT M,DEHAIS P,THIJIS G,et al.PlantCARE,a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences[J].Nucleic Acids Res,2002,30(1):325 -327.
[9] NOGUEIRA F T,CHITWOOD D H,MADI S,et al.Regulation of small RNA accumulation in the maize shoot apex[J].PLoS Genet,2009,5(1):e1000320.
[10] CHUCK G,CIGAN A M,SAETEURN K,et al.The heterochronic maize mutant corngrassl results from overexpression of a tandem microRNA [J].Nature genetics,2007,39(4):544-549.
[11] CHUCK G,MEELEY R,IRISH E,et al.The maize tasselseed4 microRNA controls sex determination and meristem cell fate by targeting tasselseed6/indeterminate spikeletl[J].Nature genetics,2007b,39(12):1517 -1521.
[12]PHILLIPS J R,DALMAY T,BARTELS D.The role of small RNAs in abiotic stress[J].FEBS letters,2007,581:3592-3597.
[13] SUNKAR R,ZHOU X,ZHENG Y,et al.Identification of novel and candidate miRNAs in rice by high throughput sequencing[J].BMC Plant Biol,2008,8:25.
[14] LU S,SUN Y H,SHI R,et al.Novel and mechanical stress-responsive microRNAs in Populus trichocarpa that are absent from Arabidopsis[J].Plant cell,2005,17:2186-2203.
[15] DING D,ZHANG L,WANG H,et al.Differential expression of miRNAs in response to salt stress in maize roots[J].Ann Bot,2009,103(1):29 -38.
[16] MICA E,GIAFRANCESCHI L,PE M E.Characterization of five microRNA families in maize[J].J Exp Bot,2006,57(11):2601 -2612.
猜你喜欢
中学生天地(A版)(2023年1期)2023-02-17
今日农业(2021年11期)2021-08-13
中西医结合肝病杂志(2020年2期)2020-10-27
北京农学院学报(2019年1期)2019-02-22
中成药(2018年7期)2018-08-04
生命科学研究(2018年1期)2018-05-29
河南农业科学(2017年4期)2017-04-12
上海农业学报(2017年3期)2017-04-10
西南农业学报(2016年5期)2016-05-17
西南农业学报(2016年6期)2016-04-16

- 河南农业大学学报的其它文章
- 玉米自交系许178背景的综3染色体单片段代换系的构建