
桑树(川桑Morus notabilis)全基因组测序*
2013-01-18 06:46桑树基因组研究团队
蚕学通讯 2013年4期
桑树基因组研究团队
(家蚕基因组生物学国家重点实验室,重庆 400716)
桑树为落叶乔木,对家蚕而言是一种重要经济饲料作物。桑叶饲养家蚕用于生产贵重蚕丝的过程至少起始于5 000年以前[1],通过“丝绸之路”塑造了世界历史。
桑科包含了37个属,约1 100个种,其中包括大家熟知的桑树、面包树、无花果树、榕树、见血封喉树等[2]。桑树属于桑属,桑属包含了10到13个公认的种,超过1 000个栽培品种[3]。这些栽培品种广泛的种植于欧亚大陆、非洲以及美国。在中国和印度,分别有约626 000、280 000hm2的土地用来栽桑养蚕[4]。由于美味的桑椹、造纸用的桑皮及其在东方传统医学方面的多种用途等特性,桑树的栽植对农民具有很强的吸引力[5-6]。
家蚕是一个专食桑叶的昆虫,为鳞翅目的模式昆虫。目前已知的大多数鳞翅目昆虫都是农业及林业上的害虫,因此对经济具有重要的影响。促摄食物质对理解植物-昆虫相互之间关系具有关键的作用,家蚕成功驯化使得深入研究这些促摄食物质成为可能,这对理解植物与昆虫之间关系是非常关键的。家蚕基因组精细图已于2008年测序完成[7-8]。然而,桑属中物种几乎没有任何基因组信息。桑树基因组测序完成不仅能够促进桑树改良,而且通过对桑树与家蚕基因组的分析将进一步加深我们对植物-植食性昆虫之间适应性进化的理解。
本文我们报道了桑树(川桑)基因组测序草图。桑树基因组估算为357MB,包含了7对染色体,通过Illumina技术测序覆盖深度达到236倍。基于组装完成的330MB基因组,鉴定到128MB的重复序列以及29 338个蛋白编码基因。比较基因组分析表明相较于其他已测序的蔷薇目植物而言,桑树进化速度更快。对桑树抗性基因的鉴定及分析将加速桑树品种改良。另外,我们在家蚕两个组织中发现了桑树的miRNA,这个结果从分子水平上暗示了植物-植食性昆虫之间可能的相互关系。
1 结 果
1.1 基因组测序和组装
川桑体细胞包含7对不同的染色体(图1),我们通过全基因组鸟枪法策略对川桑进行测序。合计78.34billion高质量的碱基(覆盖深度236倍)组装成330.79MB的桑树基因组。Scaffold N50长度为390 115bp,重叠群N50长度为34 476bp(表1、附表1和附表2)。组装完成的基因组中包含16 281kb(4.9%)的gaps以及314 510kb(95.1%)的无缺口连续序列。我们从500bp插入片段文库中选择了10.46Gb的高质量reads用来计算K-mer深度(此处选择17bp)的分布。总共得到8 577 674 309的17-mer,据此估计川桑的基因组为357.4MB(附方法、附图1和附表3)。681个scaffolds覆盖了超过80%的组装序列,其中最长的scaffold为3 477 367bp,93.96%的碱基覆盖深度超过20reads(附图2),在随机选择的10 000个表达序列标签中,97%的表达序列标签长度的90%被一个scaffold覆盖(附图4)。桑树基因组GC含量为35.02%,与其他双子叶植物GC含量类似(附方法和附图3)。

图1 川桑(M.notabilis)染色体细胞学分析
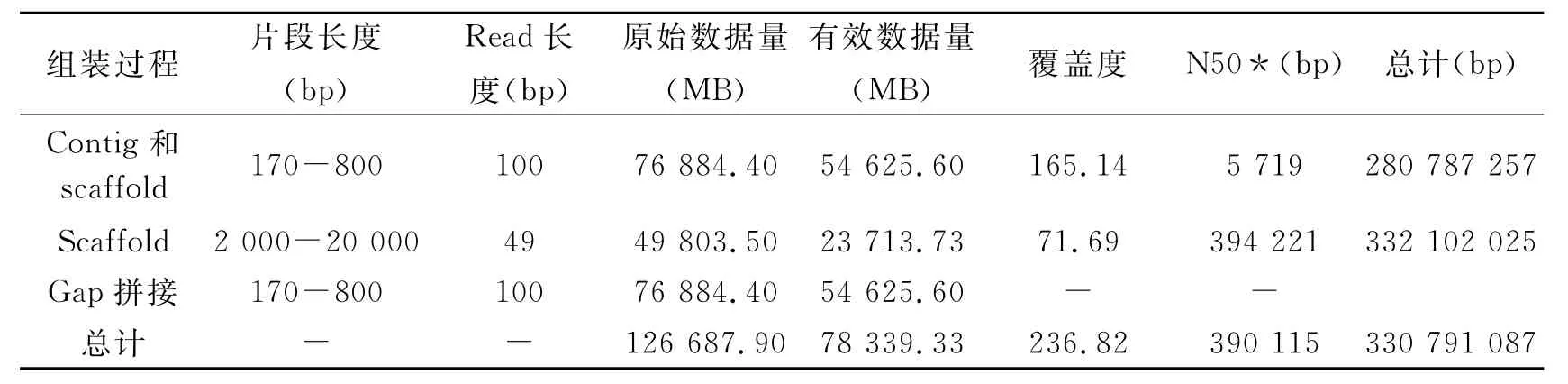
表1 桑树基因组测序及组装数据统计
1.2 重复序列
在无缺口的桑树基因组中结合从头预测以及基于Repbase文库同源搜索的方法共得到了127.98MB的重复序列(附表5)。由于从头测序技术在处理重复序列方面固有的限制,桑树基因组中转座元件的含量有可能被低估。在桑树基因组中根据平均覆盖深度、总reads所覆盖到重复序列(约127.7MB)以及非重复序列区域(约166.0MB),排除未测出的碱基(N)后,我们估计在未组装完的序列中约有18.48MB的重复序列。因此,在桑树基因组中存在约有47%的重复序列。这个比例与苹果中重复序列的比例(42%)比较接近,略高于杨树(35%)。超过50%的桑树重复序列可以划分到已知的类别中,如Gypsy-like(6.58%)以及Copia-like(6.84%)LTR型(长末端重复)转座元件。大约99.11%的转座元件有超过10%的分化度,这个结果暗示了大多数的桑树转座元件是比较古老的(附图4)。
1.3 基因预测及功能注释
结合5个组织共计21GB的转录组数据以及5 833个唯一的表达序列标签对基因模型进行预测及验证,我们在桑树基因组中鉴定到27 085个高置信度、具有完整基因结构的蛋白编码基因(附方法及附表6)。在这27 085个基因中,从头基因预测支持其中99.93%的基因,转录组测序及表达序列标签支持其中58.38%(15 811)的基因,基于同源方法搜索支持其中69.96%(18 943)的基因。这三种方法同时支持的有超过一半(52.19%)的基因。包括2 253个通过转录组测序以及表达序列标签注释得到的基因在内,我们共预测到29 338个基因(附表7)。这些基因的平均mRNA长度为2 849bp,平均编码基因长度为1 156bp,平均有4.6个外显子(附表8)。在这些基因中,转录组测序支持其中60.8%的基因,76.92%(22566/29338)的基因在功能数据库中具有同源的靶基因,如NCBI Nr(NCBI非冗余蛋白质数据库),Swissprot,InterPro,KEGG(京都基因和基因组百科全书)和COG(直系同源基因蔟)等数据库(附表9)。
基于转录组数据,我们计算了组织特异性指数τ以筛选组织特异性基因和看家基因。我们发现241、213、285、360和404个在根、皮、冬芽、雄花和叶中特异表达基因。同时,我们发现1 805个基因在这5个组织当中持续表达,其中包括116个编码核糖体蛋白和26个编码转录起始因子的基因(附图5)。
1.4 基因组进化
通过比较桑树与蔷薇目(大麻[9]、苹果[10]和草莓[11])基因组序列,可以为了解这一重要种群DNA水平上的差异性提供深刻见解。通过对桑树和12个已测序的植物(图2)单拷贝基因的系统发生分析表明桑科与蔷薇科的亲缘关系最近[12-13]。这个结果同时表明桑树和大麻两个物种的分化是在63.5百万年前,桑树和苹果两个物种的分化是在88.2百万年前,桑树和苜蓿的分化则是在101.6百万年前[14]。同义替换分布说明桑树(桑科)与大麻的分化时间比桑树与蔷薇科中苹果和草莓的分化时间晚(图3)。
我们用几种植物中不同的基因群来构建了3个系统发生树。首先,桑树中的单拷贝基因以及这些单拷贝基因在其他物种中最佳匹配的基因用来构建系统发生树(图4a)。其次,用genewise预测的单拷贝基因构建系统发生树(图4b)。再次,用不同基因组中共线性位置中的最佳匹配的序列构建系统发生树。在所有构建的系统发生树中,桑树分支比其他物种的分支长,这个结果表明桑树比其他蔷薇目物种的进化速度约快3倍。
在没有可用的遗传图谱的情况下,我们使用基于计算机的基因标记及基因组组装方法来比较桑树与草莓基因组的共线性及进化关系[15]。桑树和草莓保守共线性区域基因的分布密度是依据滑动窗口的方法计算并且以热度图的方式进行可视化(图5,附数据集1)。
最初通过对葡萄基因组的分析发现了双子叶植物普遍存在三倍化事件[16],这里我们将桑树scaffolds与桑树和葡萄最佳匹配的染色体区域进行比对可以发现另外两个不太明显的同源区域,这个结果暗示桑树同样经历了双子叶植物常见的三倍化事件。草莓和大麻中也发现一个主要的和两个次要的与葡萄具有同源性的区域(图6b,c)。草莓中的这个结果与早期报道草莓中无古多倍化事件的结果相反。基因组内和基因组间共线性区域内同源基因的同义核苷酸替换率分布进一步证明桑树、草莓和大麻经历了泛双子叶植物的六倍化事件(图6d,c)。
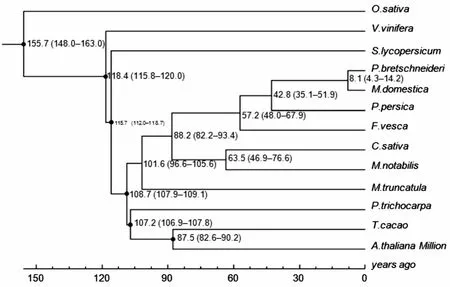
图2 13种植物的系统发生关系分析

图3 Ks分布图
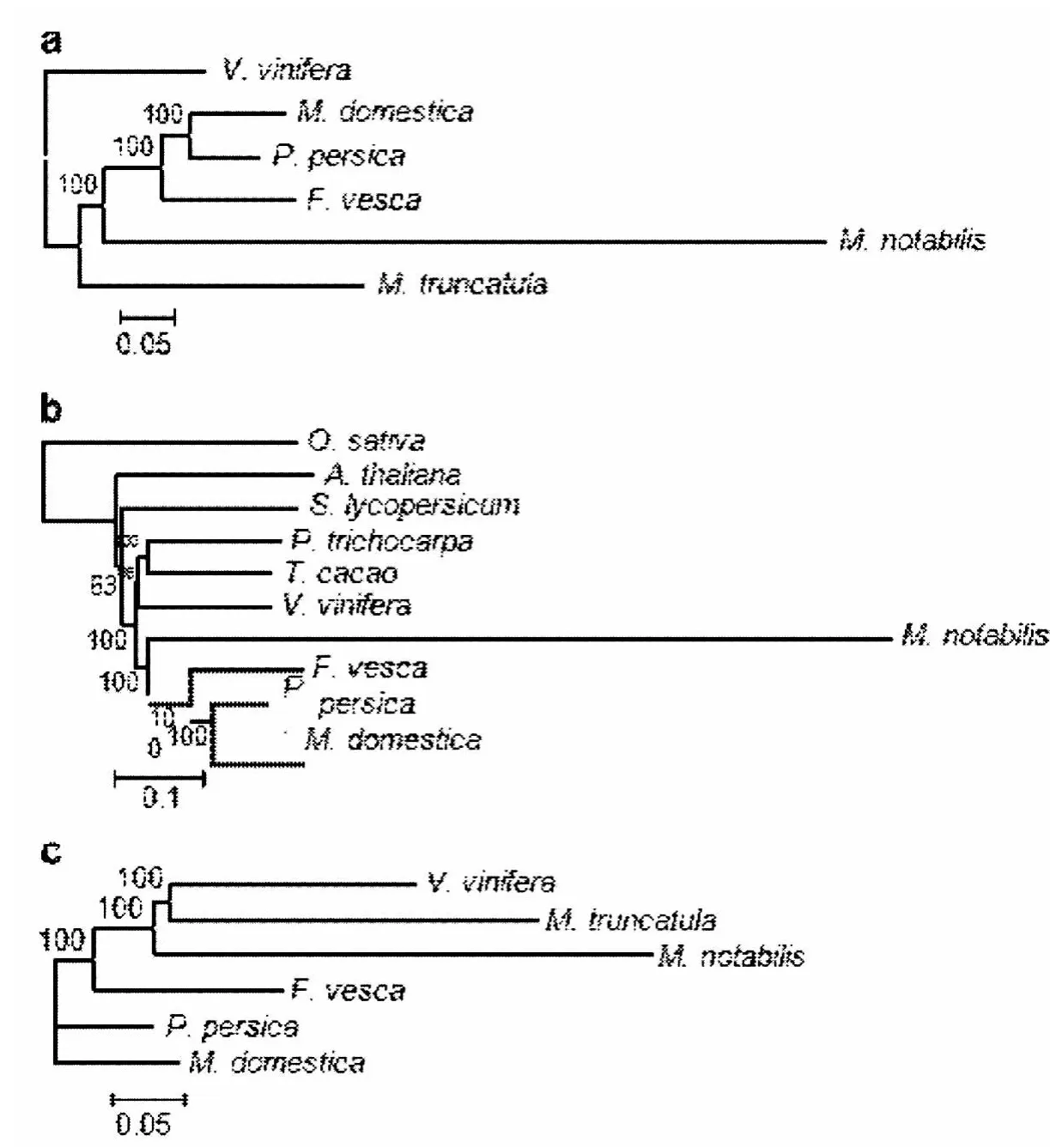
图4 川桑与其他植物的系统发生树

图5 基于草莓基因组,利用电脑模拟了川桑的基因模型
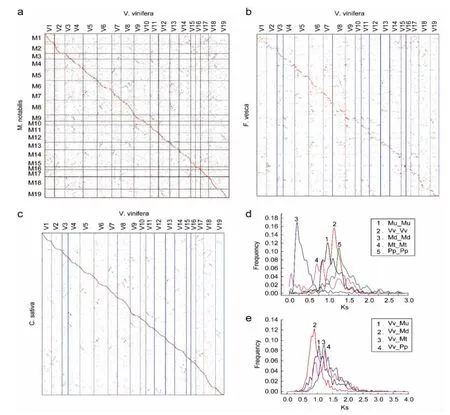
图6 物种之间的点图和Ks分布
1.5 分化选择
多种蔷薇目栽培种中不同的形态和植物化学性质可能反映了同源基因的分化选择。通过对ω(非同义核苷酸替换率(Ka)对同义核苷酸替换率(Ks)的比值(Ka/Ks))和Ks值的回归分析,在桑树-大麻、桑树-草莓、桑树-苹果和桑树-苜蓿之间鉴定出307、338、353和197个分化性选择基因对(附数据集2),其ω显著高于平均值。有趣的是,用更加严格的Fisher精确性检验进行分析发现,桑树-大麻的222对分化性选择基因(附图6和附表10)主要集中在老龄化和压力应答相关方面,这可能与植物间生命期长短不同有关。桑树-草莓,桑树-苹果的228和258对分化性选择的同源基因可能与不同的功能相关,例如Morus000754(桑树)-MDP0000252168(苹果)和Morus009486(桑树)-MDP0000290357(苹果)涉及角皮质的合成过程,可能与苹果表皮厚度有关(尽管表皮生物合成机制尚不清楚)。在桑树-蔷薇目(苹果,草莓)中受到分化性选择特别突出的基因对是与质体构成有关的(附数据集3和4),推测核酮糖二磷酸羟化酶和许多质体基因受到正向的分化选择。
1.6 抗性基因
桑树基因组有142个具有核苷酸结合位点(NBS)的抗性基因,占桑树所有基因的0.53%,与拟南芥(0.52%)和草莓(0.58%)中所占比例相当,但是低于杨树(0.86%)和苹果(1.49%)(附数据集5,附表11)。所有的 R 基因分成6个类群:TIR-NBS-LRR、CC-NBS-LRR、NBSLRR、NBS、CC-NBS以及 TIR-NBS,其中数目最多的类群为CC-NBS-LRR,包括46个基因。桑树基因组中含有127个半胱氨酸蛋白酶(CP;0.47%)以及129个天冬氨酸蛋白酶(AP;0.48%)的编码基因,与苹果(0.59%,0.37%)和草莓(0.49%,0.53%)基因组中其所占比例相当(附数据集6,7和附表12)。其中很突出的一点是有13个CP和4个AP基因在桑树的乳汁管中表达(附表13)。有趣的是,四个AP基因中的一个(Morus008067)相对苹果中其同源基因(MDP0000201076;附数据集2)而言受到分化性选择。
1.7 蛋白酶抑制子基因
为了减轻昆虫的侵袭损害,植物进化出了一种通过表达植物蛋白酶抑制子(PIs)干涉昆虫消化系统的防御机制。在已知的PI序列和它们的保守结构域的基础上,我们在桑树中鉴定了79个PIs(附表4)。在桑树基因组中注释出22个C1家族的半胱氨酸蛋白酶抑制剂基因和19个A1/C1家族的丝氨酸蛋白酶抑制剂基因,占被鉴定的蛋白酶抑制剂基因的一半。
1.8 家蚕中鉴定到的桑树miRNAs
家蚕对桑叶季节性生长的适应可能涉及跨界的分子信号。通过桑树基因组与多个植物small RNA数据库的比对,我们预测得到311个小核RNAs和233个miRNAs(附表15)。其中有5个在桑树基因组中存在,但在家蚕基因组中不存在的miRNAs,出现在家蚕幼虫血淋巴(2),前中部丝腺(2)和后部丝腺(1)(附表16)。采用另一批家蚕血淋巴对其进行重复测序,验证了家蚕血淋巴中存在桑树来源的miRNAs。
2 讨 论
早期的研究推测桑树的基础染色体数目是1419,尽管之后对两种印度桑的细胞学实验表明桑树的染色体基数有可能是7[20],但是人们一直认为桑树的染色体基数是14[19],并在大量文章引用该观点。该属植物的多倍性反映在其染色体数目的广泛性上:如川桑(M.notabilis)14条[21],印度桑(M.indica)、白桑(M.alba)28条,山桑(M.bombycis)42条,黑桑(M.nigra)308条[22]。由于多倍体基因组的复杂度高,所以选用14条染色体的川桑进行全基因组测序分析。为了证实川桑染色体数目,对处于有丝分裂中期的顶芽细胞进行细胞学分析,确认了川桑染色体数目为14条。染色体核型分析表明,川桑的14条染色体能清晰地组成7对,该结果支持印度桑关于桑树染色体基数为7的观点[20]。
系统进化分析法表明桑树与其它蔷薇目的物种聚为一个进化支。桑科按照惯例认为是属于荨麻目,而荨麻目被认为是与蔷薇目进化关系最近的一个目。然而最近的一个研究认为榆科、大麻科、桑科和荨麻科属于单一的进化支[23],将其命名为urticalean rosids[24]。随后被子植物种系发生群III将桑科归属为蔷薇目[13],本研究也支持这种重新分类的结果。
桑树在核酸水平上进化速度很快,其快速进化的基因可能使桑树不仅对本地环境有很好的适应性,还能促进其传播到欧洲,非洲和美国。与桑树快速变化的核酸水平相比,蔷薇目的倍性进化则非常保守。桑树,草莓,大麻,番木瓜和葡萄都经历过最近的泛双子叶植物六倍化事件。桑树的染色体数目高达308条(44×)[22],草莓高达70条,广泛存在的高倍数性揭示了这些谱系能接受新一轮多倍化带来的益处。
桑树是一种多年生的木本植物,对桑树持续剪伐不仅能采集桑叶养蚕,还能提高桑叶产量,但是剪伐会增加桑树受到害虫侵扰和病原菌侵染的风险,因此,桑树需要强大的防御系统抵挡剪伐带来的生物胁迫。R基因编码的蛋白能识别病原菌的效应子,比如其相应的无病毒的基因产物[25]。关于植物R基因的研究大多数集中在包含NBS结构域的R基因上[26]。在桑树基因组中我们共鉴定到142个含NBS结构域的R基因。桑树是一种含有乳汁的植物,乳汁中的蛋白组分,比如几丁质酶样蛋白涉及抵御微生物或植食性昆虫的侵扰[27-29]。番木瓜乳汁管中的半胱氨酸蛋白酶和猪笼草的天门冬氨酸蛋白酶对植食性昆虫均有毒性[30-31]。通过全基因组分析发现桑树有127个半胱氨酸蛋白酶编码基因和129个天冬氨酸蛋白酶编码基因,这些基因的功能研究将会扩展我们对桑树防御机制的理解。
植物防御机制能干扰昆虫的消化系统,关于寡食性家蚕是如何绕过桑树的防御机制,这个问题至今为止仍不清楚。植物蛋白酶抑制剂能降低植食性昆虫中肠消化酶的活性,从而导致昆虫发育严重畸形,死亡,生殖率降低[32-33]。早期研究报道了植物产生的多结构域、多聚体结构的蛋白酶抑制剂对斜纹夜蛾具有抗营养作用[34]。昆虫可以通过诱导产生一些对蛋白酶抑制剂不敏感的蛋白酶以及用体内特殊的蛋白酶降解植物的蛋白酶抑制剂等方法来避开植物蛋白酶抑制剂的作用[35-36]。一种对十字花科有害的著名鳞翅目昆虫—小菜蛾能使芥末的胰蛋白酶抑制剂2失活,从而打破宿主植物的抵御[37]。受益于家蚕和桑树的基因组序列,对其转录组的比较分析能够推进我们对植物-植食性昆虫相互适应关系的理解。
在家蚕三个组织的小RNA测序数据中发现五个来源于桑树而非家蚕的miRNA,其中的MIR156在水稻营养生长期的老叶中高量表达,该miRNA在植物从幼态向成熟态的转变中起主要作用[38-40]。同时我们注意到水稻的MIR168a能转运到人体内,并调节低密度脂蛋白受体衔接蛋白1的表达水平[41],那么存在家蚕丝腺中的桑树MIR156是否向家蚕发出桑叶衰老的信号,促进家蚕吐丝成茧,亦或是家蚕组织特异存在的其它桑树miRNA在家蚕的发育中是否起作用,这些问题至今仍不清楚。
综上所诉,基因组信息对现代桑树遗传研究是十分重要的资源。桑树基因组的特点,如基因家族,片段重复,共线性区域等不仅丰富了植物比较基因组可用的数据,并且促进了与桑科密切相关物种靶基因的鉴定。基于基因组序列发展的遗传标记可以用于构建遗传图谱,定位克隆,品系鉴定以及标记辅助的筛选。这些分子工具和基因组技术的应用将会有效促进农业的发展。桑树和家蚕作为植物-植食性动物相互作用的模式系统,二者的基因组序列对于更深层次解析普遍存在大多数陆生环境中的生物学合伙关系提供了一个独一无二的机会。
3 研究方法
3.1 川桑核型分析
川桑嫩叶用2mM 8-羟基喹啉在室温下处理3h,之后用3:1的甲醇:冰醋酸在4℃固定2h。固定好的叶片用1/15MKCl溶液处理30min,随后用2.5%(W/V)的纤维素酶和2.5%(W/V)的果胶酶(YaKult Co.,日本)在37℃消化1.5h。消化后的叶片用ddH2O处理10min,之后再用3:1的甲醇:冰醋酸在室温下固定30min。再固定的叶片打碎后滴两滴悬浮液在载玻片上,室温下用吉姆萨染液染色6小时,随后在显微镜下进行观察(Olympus,日本)。
3.2 DNA和RNA制备
测序用桑树品种为野生桑种川桑,其含有14条染色体。川桑冬芽的基因组DNA用CTAB法进行提取并用于测序文库的构建。五个组织(根、一年生皮、冬芽、雄花和叶)的总RNA参照Wan和Wilhins等的方法进行提取[42],并用不含RNA酶的DNA酶Ⅰ(New England BioLabs)在37℃下消化30min以去除剩余的DNA。用含有oligo(dT)的珠子分离含有poly(A)的mRNA。用随机的六聚引物和反转录酶(Invitrogen)合成cDNA第一链。用DNA聚合酶(New England BioLabs)和RNA酶H(Invitrogen)合成cDNA第二链。
3.3 基因组测序
桑树基因组测序使用全基因组鸟枪法。测序文库根据制造商(Illumina,圣地亚哥,加拿大)的说明进行制备。对于短插入片段的DNA文库,将5μg基因组DNA用压缩氮气喷雾法进行片段化处理。片段化的DNA末端用碱基A进行钝化处理,然后将3’末端含有一个碱基T的DNA接头(Illumina)与DNA片段进行连接。之后用2%琼脂糖凝胶将连接产物进行分离,通过切胶纯化各个插入大小的DNA片段。对于长的mate-paired文库(大于等于2Kb),将10~30μg基因组DNA用压缩氮气喷雾法进行片段化处理,然后用生物素标记的dNTPs对片段进行处理,通过凝胶选择2Kb,5Kb和10Kb的主要条带。之后DNA片段通过自连而环化。将DNA片段的两端进行结合并用核酸外切酶对线性DNA片段进行消化。消化之后环化的DNA片段重新进行片段化,随后利用生物素与抗生蛋白链菌素的相互作用,用磁珠富集含有之前结合的末端的DNA,之后将这些末端进行钝化并加上含有碱基A的接头。我们根据制造商的说明进行了如下的paired-end(PE)测序流程:成蔟、模板杂交、等温扩增、线性化、封闭以及测序引物的变性和杂交。测序完成后用base-calling程序(SolexaPipeling-0.3 )从最初的荧光图像中获得序列。
3.4 基因组组装
在进行从头组装之前,我们通过以下五个步骤对低质量的数据进行过滤:(1)根据Hiseq2000的测序质量报告去除5’和3’末端的低质量的碱基;(2)去除含有N大于10%的序列;(3)去除低质量碱基(Q<8)大于50%的序列;(4)去除被接头污染的序列;(5)去除在文库构建过程中因PCR引起的重复的序列。SOAPdenovo是由深圳华大基因研发的一种基因组组装软件,该软件采用基于图论的算法和分步组装的策略[43]。我们首先用49-kmers对小插入片段的文库(<1kb)进行组装。然后我们用41-kmers将所有过滤得到的序列重新比对到组装好的重叠群上并且将比对上的序列编译为可用的重叠群。根据paired-end信息,我们共采用了七步,从170bp的插入文库到20kb的插入文库,将重叠群组装成scaffold。为了填补scaffold中的空隙,我们收集了paired-end的序列,如果其中一端比对到一个重叠群上而另一端位于空隙中,则重复进行一次局部的组装。
3.5 转座元件(TE)和重复序列
为了预测桑树基因组中的TE,我们首先用RepeatModeler(version 1.0.3,http://www.repeatmasker.org/RepeatModeler.html),RepeatScout[44](version 1.0.5,http://bix.ucsd.edu/repeatscout/)和Piler[45](version 1.0,http://www.drive5.com/piler/)构建了一个 TE库,之后用 RepeatMasker[46](version 3.2.9,http://www.repeatmasker.org/)对桑树基因组中的TE进行了一个从头预测。我们同时也使用RepeatMasker和ProteinMask(version 3.0)对已知类型的TE进行了预测,在预测过程中我们使用的TE库包括Repbase[47](version 15.02,http://www.girinst.org/repbase/)和TIGR中的双子叶植物 TE库[48](version 3.0,ht-tp://plantta.jcvi.org)。串联重复序列使用Tandem Repeats Finder(TRF,version 4.04,http://tandem.bu.edu/trf/trf.html)来进行预测。简单重复序列、微卫星序列和低复杂度重复序列使用RepeatMasker进行预测(使用“-noint”选项)[49]。将桑树基因组中已进行分类的TE家族与Repbase(v15.02)中的共有序列进行比对以确定这些TE的序列分化度。
3.6 基因预测和注释
桑树基因预测共使用了三种方法:基于同源性比较的方法、从头预测方法和基于表达序列标签(EST)/转录组数据的方法。其中高置信度的基因是用前两种方法预测得到。为了注释桑树中的蛋白编码基因,我们将其27085个高置信度基因的核苷酸序列与NCBI、KEGG、COG和Swissport等数据库进行比对,比对的阈值设为1e-5。蛋白质结构域和功能预测在Iprscan(v4.4.1)上进行注释。
3.7 RNA和EST测序
cDNA文库的制备和测序参照Illumina的操作流程。用TopHat(v1.3.3)将RNA测序得到的序列与桑树基因组进行比对。通过计算每百万条reads中覆盖到每kb上的reads数来衡量五个组织中的基因表达水平,并且通过计算组织特异性的指数τ来鉴定组织特异性表达的基因。对于EST测序,将相同的五个组织的RNA混合并用Creator SMART cDNA Kit(Clontech)进行cDNA的合成。用Trimmer-Director kit(Evrogen)构建标准化的cDNA文库。从该文库中随机挑选10 000个克隆用ABI3730(Applied Biosystem)进行测序。
3.8 非编码RNA基因
桑树基因组中的tRNA使用tRNAscan-SE(v1.23)软件进行预测(使用“eukaryotes”选项)[50]。用BLASTN将桑树基因组与植物rRNA序列进行比对(阈值为1e-5),序列一致性大于85%并且高分值片段对的长度大于50bp的结果认为是桑树的rRNA。对于microRNA和snRNA,首先用BLASTN将桑树基因组与Rfam数据库(v9.1)进行比对(阈值为1),比对结果进一步用INFERNAL软件进行分析,该软件根据RNA的结构和序列相似性对microRNA和snRNA进行预测。
3.9 模拟基因染色
我们通过BLASTP比对(阈值为1e-5)鉴定桑树和草莓之间的直系同源基因对,得到的结果进一步用Mcscan分析以鉴定两个物种间的共线性区域。通过使用Genome Zipper软件,根据草莓的连锁群,将桑树中与之对用的共线性区域的scaffold进行连接。计算每500kb滑动窗口内的基因密度和对应的直系同源基因的密度并将结果分布做热度图。
3.10 在家蚕的组织中鉴定桑树的miRNAs
用mirVana PARIS kit(Ambion,美国)从12ml的家蚕血淋巴(取自五龄5天幼虫)中提取小RNA。提取的小RNA测序参照Liu等人的操作流程进行[51]。前中部和后部丝腺的小RNA数据从http://www.ncbi.nlm.nih.gov/gds?term=GSE17965上下载。这三个家蚕组织的小RNA序列通过BLASTN与桑树预测的miRNAs进行比对(完全匹配)以鉴定家蚕组织中存在的桑树来源的miRNAs。
3.11 系统发生树的构建和物种形成时间的确定
基于最大似然法,我们利用13个植物物种的单拷贝基因构建了系统发生树。通过BLAST比对(阈值为1e-10)确定物种间的直系同源基因对,它们之间的同义替换率(Ks)[52]使用PAML软件包中的yn00程序进行计算[53]。基于Ks值的物种形成时间根据以下公式计算:T= Ks/2λ(其中λ= 6.1×10-9)[54]。根据非同义替换率(Ka)和 Ks之间的回归分析(95%预测区间范围)[55]推测桑树和其他四种植物间可能受到正选择的直系同源基因对。ω值大于预测区间上限的基因对认为是受到正选择的。用BLAST2GO[56]对具有高ω值的基因对的基因本体论类群进行预测,其结果用Fisher精确性检验进行验证(P值<0.05)。
3.12 推测基因共线性
我们用MCSCAN57(一种多染色体比对工具)和COLINEARSCAN58(一种成对染色体比对工具)相结合来推测共线性基因,这些共线性基因用于系统发生和进化分析。
3.13 推测进化事件的时间
我们利用物种间的共线性基因和物种内高度可信的同源基因来推测进化事件。例如,桑树scaffold之间的共线性基因可能来源于祖先的多倍化事件(如果存在的话),而桑树和葡萄之间的共线性基因可能来源于两个物种的分化。Ks使用PAML[53]中的Nei-Gojobori方法[52]计算。通过描绘Ks值的分布来推测进化事件的相对时间。
3.14 同源基因点图
在这一部分分析中我们使用之前介绍的预测基因集和Genewise[59]预测的一个基因集。葡萄、苹果、草莓和大麻的基因组序列和注释下载自在线数据库,所有的数据使用2012年10月之前的最新版本。在比较有可用的拟染色体的基因组时,我们用BLASTP进行蛋白-蛋白之间的比对,从而找到可能的同源基因,得到的结果用于作点图;作图过程中以基因在染色体上的顺序为坐标。当比较没有可用的拟染色体的基因组时(例如桑树和大麻),我们选择一个有可用的拟染色体物种(例如葡萄)的编码基因序列,用BLASTN与桑树和大麻的基因组进行比对,比对的结果用于作点图。为了推测基因组重复事件,没有锚定的scaffold根据它们在葡萄基因组上的最好匹配区域连接成假定的拟染色体,桑树和大麻假定的拟染色体就是用这种方法构建。在点图中,相应的葡萄的基因区域将对应两个匹配的区域。
3.15 本研究中所使用的数据
本研究中所使用的基因组数据从以下网址中下载,并列出了其在NCBI上的登录号。拟南 芥 (TAIR9):ftp://ftp.arabidopsis.org/Genes/TAIR9_genome_release/,GCA _000001735.1。大麻:http://genome.ccbr.utoronto.ca/downloads.html,GCA_000230575.1。番木 瓜 (version 1th):ftp://ftp.jgi-psf.org/pub/compgen/phytozome/v5.0/Cpapaya/,GCA_000150535.1。黄瓜(version 1th):http://cucumber.genomics.org.cn/page/cucumber/download.jsp,GCA_000004075.1。草莓(version 1.1):http://www.rosaceae.org/species/fragaria/fragaria_vesca/genome_v1.1,GCA_000184155.1。大豆(version 1.0):ftp://ftp.jgi-psf.org/pub/compgen/phytozome/v5.0/Gmax/,GCA_000004515.1。苹果(version 1.0):http://genomics.research.iasma.it/index.html,GCA_000148765.2。苜蓿:ftp://ftp.jgi-psf.org/pub/compgen/phytozome/v8.0/Mtruncatula/,GCA_000219495.1。杨树 (version 5.0):ftp://ftp.jgi-psf.org/pub/compgen/phytozome/v5.0/Ptrichocarpa/,GCA _000002775.1。桃:ftp://ftp.jgi-psf.org/pub/compgen/phytozome/v8.0/Ppersica/,GCA_000346465.1。白梨:http://peargenome.njau.edu.cn:8004/default.asp?d=1&m=1,GCA_000315295.1。可可(version 1.0):http://cocoagendb.cirad.fr/gbrowse/download.html,GCA_000403535.1。葡萄:http://www.genoscope.cns.fr/externe/Download/Projets/Projet_ML/data/12X/,GCA_000003745.2。
论 文 链 接:http://www.nature.com/ncomms/2013/130919/ncomms3445/full/ncomms3445.html.
猜你喜欢
四川蚕业(2022年2期)2022-11-19
音乐教育与创作(2022年1期)2022-04-26
四川蚕业(2021年2期)2021-03-09
四川蚕业(2021年1期)2021-02-12
今日农业(2020年16期)2020-12-14
文苑(2018年22期)2018-11-19
小学生作文(中高年级适用)(2018年3期)2018-04-18
新农业(2016年18期)2016-08-16
学生天地·初中(2016年6期)2016-05-14
西南军医(2016年6期)2016-01-23
