
Quantification of sibutramine and its two metabolites in human plasma by LC-ESI-MS/MS and its application in a bioequivalence study
2012-12-23 04:58VenktSureshPonnuruChllRmRoNdendl
Venkt Suresh Ponnuru, B.R. Chll, RmRo Ndendl
aChalapathi Institute of Pharmaceutical Sciences, Lam, Guntur 522034, Andhra pradesh, India
bNirmala College of Pharmacy, Kadapa 516002, Andhra pradesh, India
cKrishna University, Machilipatnam 521001, Andhra pradesh, India
1. Introduction
Sibutramine hydrochloride monohydrate [(±)-dimethyl-1-[1-(4-chlorophenyl)cyclobutyl]-N,N,3-trimethylbutan-1-amine hydrochloride monohydrate] is currently used for the treatment of obesity. The chemical formula of sibutramine is C17H29Cl2NO and its molecular weight is 334.33.Sibutramine acts by inhibiting the re-uptake of neurotransmitter serotonin,norepinephrine and dopamine and enhancing satiety. Consequently, it prevents compulsive eating and inhibits the sensation of hunger in obese patients [1,2]. Today obesity can be considered as a chronic illness of epidemic proportion and its incidents have increased exponentially [3]. For many patients,dieting and physical exercise are not sufficient to achieve and maintain the desired weight loss.The use of anti-obesity drugs such as sibutramine is needed.
Literature survey reveals that quantification of sibutramine in pharmaceutical, synthetic/dietary supplements and herbal products was reported using LC-MS [4-7] and HPLC [8-11]in biological matrices, e.g. rats [12-18]. Only a few methods were reported for the quantification of sibutramine and its two metabolites in human plasma using LC-MS/MS [19-24].Kang et al. [21] developed a method with the linearity range of 0.1 - 0.8 ng/mL; they used imipramine as the internal standard and a more complicated solid phase extraction method to isolate sibutramine and its two metabolites. Ding et al. [22] established a method with a linearity range of 0.05-20.0 μg/L for sibutramine and N-des methyl sibutramine and 0.1-30.0 μg/L for N-di-des methyl sibutramine. They used phenyl propyl amine hydrochloride as an internal standard,liquid-liquid extraction for extraction of drug and IS and compared test and reference formulations with 18 volunteers.
Chen et al. [23] reported a method with a linearity range of 0.328-32.8 ng/mL for sibutramine, N-Des methyl sibutramine and N-di-des methyl sibutramine. They used liquid-liquid extraction and propronolol as an internal standard and compared test and reference formulations with 20 volunteers.Bae et al.[24]developed a high-sensitivity method with a good linearity range of 50-20000 pg/mL and a shorter runtime and compared the drug and metabolites with chlorphenaramine as an internal standard. However, we experienced many difficulties with the Bae's method in terms of reproducibility and stability in long run analysis.
It is important to develop a reliable bio-analytical method with a properly deuterated derivative or other analog as the internal standard to study the matrix effect and reproducibility. It is not required to consider the runtime when comparing the reproducibility and stability of analytical batches. The goal of the present study was to develop a reliable and accurate method for the quantification of sibutramine and its two metabolites with respective deuterated internal standards. Moreover, the sample extraction method should be simple and the analytical method should be highly sensitive with good linearity and the use of a small amount of plasma.
2. Materials and methods
2.1. Chemicals and reagents
Sibutramine hydrochloride (SB) was purchased from Symed labs. N-des-methyl sibutramine hydrochloride (DSB), N-didesmethyl sibutramine hydrochloride(DDSB),sibutramine d7 hydrochloride (SB d7), N-des-methyl sibutramine d7 hydrochloride (DSB d7), N-di-des methyl sibutramine d7 hydrochloride (DDSB d7) were obtained from Toronto Research Chemicals, Canada (Fig.1). Tertiary butyl methyl ether(TBME), HPLC grade methanol and acetonitrile were purchased from J.T. Baker, USA. Potassium dihydrogen phosphate (KH2PO4, reagent grade) and ammonium formate(reagent grade) were purchased from Merck Limited, Worli,Mumbai. Human plasma was obtained from Navajeevan blood bank, Hyderabad, India. Ultra pure water from MilliQ-system (Millipore) was used throughout the study.

Figure 1 Chemical structures of (a) sibutramine HCl, (b) N-des methyl sibutramine HCl, (c) N-di des methyl sibutramine HCl,(d) sibutramine d7 HCl, (e) N-des methyl sibutramine d7 HCl,(f) N-di des methyl sibutramine d7 HCl.
2.2. Instrumentation
The Agilent 1200 Series HPLC system (Agilent Technologies,Waldbronn, Germany) connected to the API 4000 triple quadrupole instrument (ABI-SCIEX, Toronto, Canada) with turbo electrospray interface in positive ionization mode was used. Data processing was performed on Analyst 1.4.1 software package (SCIEX).
2.3. Detection
The mass transitions of m/z 280.3/124.9, 266.3/125.3, 252.2/124.9, 287.3/125.2, 273.2/125.0, and 259.3/125.1 were used for the quantification of SB, DSB, DDSB, SB d7, DSB d7, and DDSB d7, respectively (Table 1).
2.4. Chromatographic conditions
Zorbax SB-C18 (4.6 mm×75 mm, 3.5 μm, 80 ˚A) analytical column was used and it was maintained at 40°C. The mobile phase consisted of 5 mM ammonium formate:acetonitrile(10:90, v/v), which was run at a flow-rate of 0.6 mL/min.The analytes and internal standards were eluted at 9.2 min(SB, SB d7), 8.2 min (DSB, DSB d7) and 6.2 min (DDSB,DDSB d7) with a total runtime of 13 min for each injection.
2.5. Calibration standards and quality control samples
Standard stock solutions of SB(100.0 μg/mL),DSB(100.0 μg/mL), DDSB (100.0 μg/mL) were prepared in methanol. From each stock solution, 500.0 ng/mL, 25.0 ng/mL, 2.5 ng/mL intermediate dilutions were prepared in plasma.Aliquots of 500.0 ng/mL,25.0 ng/mL and 2.5 ng/mL were used to spike blank human plasma in order to obtain calibration standards at 10.0, 20.0,200.0, 800.0, 1500.0, 3000.0, 4500.0, 6000.0, 7500.0 and 10,000.0 pg/mL. Four levels of quality control (QC) concentrations at 10.0, 30.0, 3500.0 and 8000.0 pg/mL (lower limit of quantification (LLOQ), low quality control (LQC), medium quality control (MQC) and high quality control (HQC)) were prepared with different plasma lot. Spiked calibration standards and quality control standards were stored at -30°C until analysis. Standard stock solutions of SB d7 (100.0 μg/mL),DSB d7 (100.0 μg/mL) and DDSB d7 (100.0 μg/mL) were prepared in methanol. SB d7, DSB d7 and DDSB d7 stock solutions were further diluted to 30.0 ng/mL (spiked concentration of internal standard) with 50% methanol and stored in the refrigerator at 2-8°C until analysis.
2.6. Sample preparation
Liquid-liquid extraction was used to extract the drug and internal standard. For this purpose, 100 μL of respective plasma sample was placed in a polypropylene tube and mixed with 50 μL of internal standard (30.0 ng/mL). This was followed by addition of 100 μL of 10 mM KH2PO4 solution and 2.5 mL of methyl tertiary butyl ether (MTBE) and vortexed approximately for 5 min. The samples were then centrifuged at 4000 rpm for 10 min at 20°C.Subsequently,the supernatant was transferred to respective polypropylene tubes and evaporated under a stream of nitrogen gas at 40°C.After drying, samples were reconstituted with 200 μL of reconstitution solution (acetonitrile:5 mM ammonium formate (90:10, v/v))and vortexed for 2 min. Finally, the samples were transferred to autosampler vials and 20 μL was injected into the LC-MS/MS.
2.7. Selectivity and specificity
Selectivity was determined by analyzing the blank human plasma samples from six different sources (donors) with an additional hemolyzed group and lipidemic group to assess interference at the retention times of analytes. The peak areas of SB, DSB and DDSB in blank samples should not be more than 20% of the mean peak area of LOQ of SB, DSB and DDSB.Similarly,peak areas of SB d7,DSB d7 and DDSB d7 in a blank sample should not be more than 5% of the mean peak area of LOQ of SB d7, DSB d7 and DDSB d7.
2.8. Precision and accuracy
Precision and accuracy were determined by replicate analysis of quality control samples (n=6) at low quality control (LQC),medium quality control(MQC)and high quality control(HQC)levels.The CV should be less than 15%and accuracy should be within 15% except LLOQ where it should be within 20%.
2.9. Matrix effect
The matrix effect caused by plasma was evaluated by examining the ion suppression/enhancement in a signal when comparing the absolute response of QC samples after pretreatment(liquid-liquid extraction with MTBE) with that of the reconstituted samples. Experiments in triplicate were performed at MQC levels with 6 different plasma lots with the acceptable precision (%CV) of ≤15%.
2.10. Recovery
The extraction efficiencies of SB,DSB,DDSB and SB d7,DSB d7,and DDSB d7 were determined by analysis of 6 replicates at each quality control concentration level for SB, DSB and DDSB and at one concentration for the internal standard SB d7,DSB d7 and DDSB d7.The percent recovery was evaluated by comparing the peak areas of extracted standards to the peak areas of non-extracted standards (blank plasma residue).
2.11. Limits of detection and quantification(LOD and LOQ)
The limit of detection (LOD) is a parameter that defines the lowest concentration in a sample that can be detected from background noise but cannot be quantitated. LOD was determined using the signal-to-noise ratio (s/n) of 3:1 by comparing the results from samples of known concentrations of analytes with blank samples. The limit of quantitation(LOQ)is defined as the lowest concentration of an analyte thatcan be determined with acceptable precision and accuracy.The LOQ was found by analyzing a set of plasma standards with known concentrations of SB, DSB and DDSB.
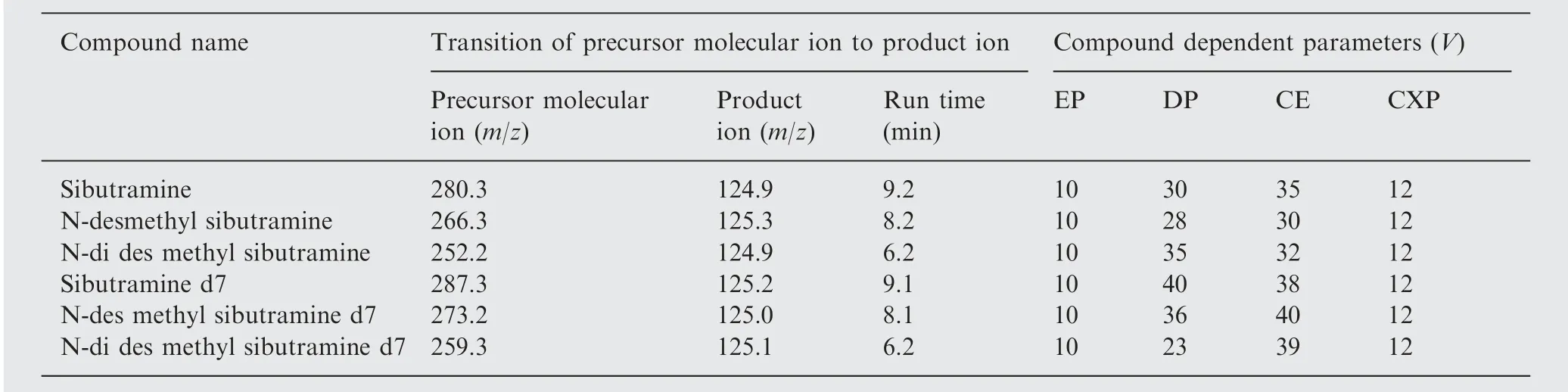
Table 1 Mass parameters of sibutramine (SB) and its two metabolites N-des methyl sibutramine (DSB), N-di des methyl sibutramine (DDSB) and sibutramine d7 (SB d7), N-des methyl sibutramine d7 (DSB d7), N-di des methyl sibutramine d7(DDSB d7).
2.12. Stability
Stock solution stability was determined by comparing the area response of analyte and internal standard in the stability samples with the area response of samples prepared from fresh stock solution. Stability studies in plasma were performed at the LQC and HQC concentration levels using six replicates at each level. Analyte was considered stable if the % change is less than 15% as per US FDA guidelines [25]. The stability of spiked human plasma samples stored at room temperature(bench top stability) was evaluated for 72 h. The stability of spiked human plasma samples stored at -30°C in autosampler (autosampler stability) was evaluated for 78 h. The autosampler sample stability was evaluated by comparing the extracted plasma samples that were injected immediately(time 0 h), with the samples that were reinjected after storing in the autosampler at 20°C for 78 h. The reinjection reproducibility was evaluated by comparing the extracted plasma samples that were injected immediately (time 0 h), with the samples that were re-injected after storing in the autosampler at 20°C for 27 h.The freeze-thaw stability study was conducted by comparing the stability samples that had been frozen at -30°C and thawed three times, with freshly spiked quality control samples.Six aliquots each of LQC and HQC concentration levels were used for the freeze-thaw stability evaluation.For long term stability evaluation, the concentrations obtained after 71 days were compared with initial concentrations.
2.13. Analysis of human plasma samples
The validated method has been successfully used to analyze SB, DSB and DDSB concentrations in the plasmas of 40 human volunteers under fasting conditions after oral administration of a single capsule containing 15 mg (1×15 mg) SB,DSB and DDSB. The study was designed as a randomized,two-period, two-sequence, two-treatment, single dose, and open label bioequivalence study. The reference formulation was used as MERIDIA Capsule(15 mg,Abbot,USA)and the test formulation was used for APL Research Pvt. Ltd (15 mg,Test capsule), India. The study was conducted according to current GCP guidelines and after the volunteer signed written consent. The study protocol was also approved by an authorized ethics committee. There were a total of 28 blood collection time points including the predose sample.The blood samples were collected at time intervals of 0, 0.33, 0.5, 0.667,0.833, 1.0, 1.25, 1.5, 1.75, 2.0, 2.5, 3.0, 3.5, 4.0, 4.5, 5, 6, 7, 8,10, 12, 16, 24, 36, 48, 60, 72 and 96 h in separate vacutainers containing K2EDTA as an anticoagulant. The plasma was separated from the blood by centrifugation at 3500 rpm, 2-8°C for 10 min,and kept at-30°C until sample analysis.Test and reference were administered separately to the same human volunteers under fasting conditions with proper washing periods as per protocol approved by IEC.
2.14. Pharmacokinetics and statistical analysis
Post analysis was conducted and the pharmacokinetic parameters were obtained using Win-Nonlin®software version 9.2. Blood samples were collected for a period of 3-5 times that of the terminal elimination half-life(t1/2).As per standard guidelines AUC0-t, AUC0-∞, Cmax,t1/2and tmaxwere calculated for SB, DSB and DDSB in plasma samples. According to regulatory guidelines AUC0-t, AUC0-∞and Cmaxvalues should be within 80-125% for test and reference products[26,27].
3. Results and discussion
3.1. Method development and validation
The goal of this work was to develop and validate a simple,rapid and sensitive method for the quantitative determination of SB, DSB and DDSB in plasma samples. LC-MS/MS has been used as one of the most powerful analytical tools in clinical pharmacokinetics for its selectivity, sensitivity and reproducibility.The MS optimization was performed by direct infusion of SB, DSB, DDSB and SB d7, DSB d7, DDSB d7 solutions into the ESI source of the mass spectrometer. The vital parameters like ionization with positive mode, temperature at 400°C, ion spray voltage at 5500 V, gas parameters such as nebulizer gas at 20 psi and heater gas at 40 psi were obtained and compound parameters like declustering potential(DP), entrance potential (EP), collision energy (CE) and collision cell exit Potential(CXP) were individually optimized(Table 1) to obtain the highest sensitivity and most efficient ionization to form the respective product ions from the protonated SB, DSB, DDSB and SB d7, DSB d7, DDSB d7 molecules(Fig.2).Chromatographic conditions,especially the composition of the mobile phase and selection of suitable column,were optimized through several trials to achieve the best resolution and highest signals of analytes and internal standards.Different extraction methods like solid phase extraction, liquidliquid extraction and precipitation methods were optimized for extraction of SB, DSB, DDSB and SB d7, DSB d7, DDSB d7 from the plasma samples. Good separation and elution were achieved using 5 mM ammonium formate:acetonitrile(10:90,v/v)as the mobile phase, at a flow-rate of 0.6 mL/min with 20 μL injection volume. Liquid-liquid extraction was chosen to recover the drug and internal standard from the plasma. The retention time was optimized at 9.2,8.2,6.2 min for SB,DSB,DDSB and 9.1, 8.1, 6.2 min for SB d7, DSB d7, DDSB d7, respectively(Fig.3).
3.2. Linearity
Calibration curve was plotted as the peak area ratio (SB/SB d7, DSB/DSB d7 and DDSB/DDSB d7) versus (SB, DSB,DDSB)concentration.Calibration was found to be linear over the concentration range of 10.0-10,000.0 pg/mL. The correlation coefficient (r2) was greater than 0.9997 for all curves(Table 2).
3.3. Selectivity
The selectivity of the method was assessed by comparing chromatograms of blank plasma. No significant endogenous peaks were observed at respective retention time of SB, DSB,DDSB and SB d7, DSB d7, DDSB d7. The results indicate that the method was specific and selective (Fig.4).
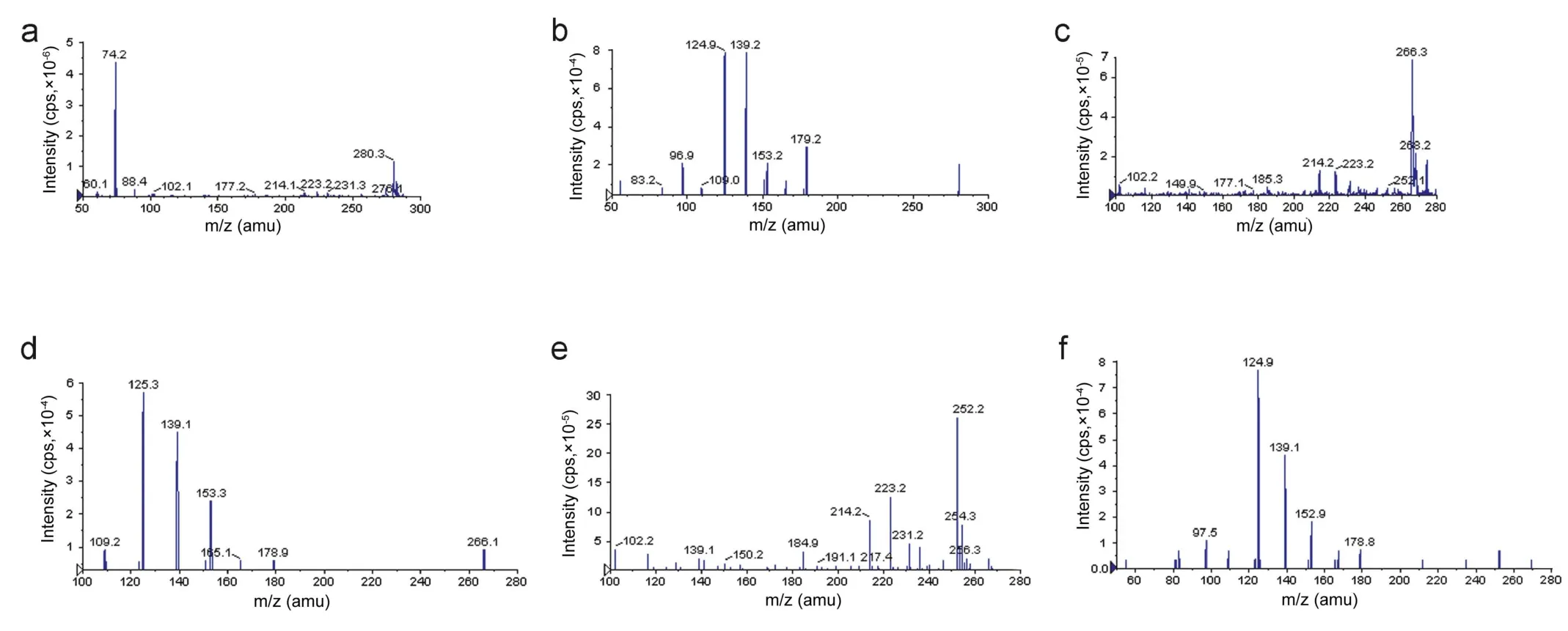
Figure 2 (a)Parent ion mass spectra of sibutramine(SB),(b)product ion mass spectra of sibutramine,(c)parent ion mass spectra of Ndes methyl sibutramine(DSB),(d)product ion mass spectra of N-des methyl sibutramine,(e)parent ion mass spectra of N-di des methyl sibutramine (DDSB), and (f) product ion mass spectra of N-di des methyl sibutramine.
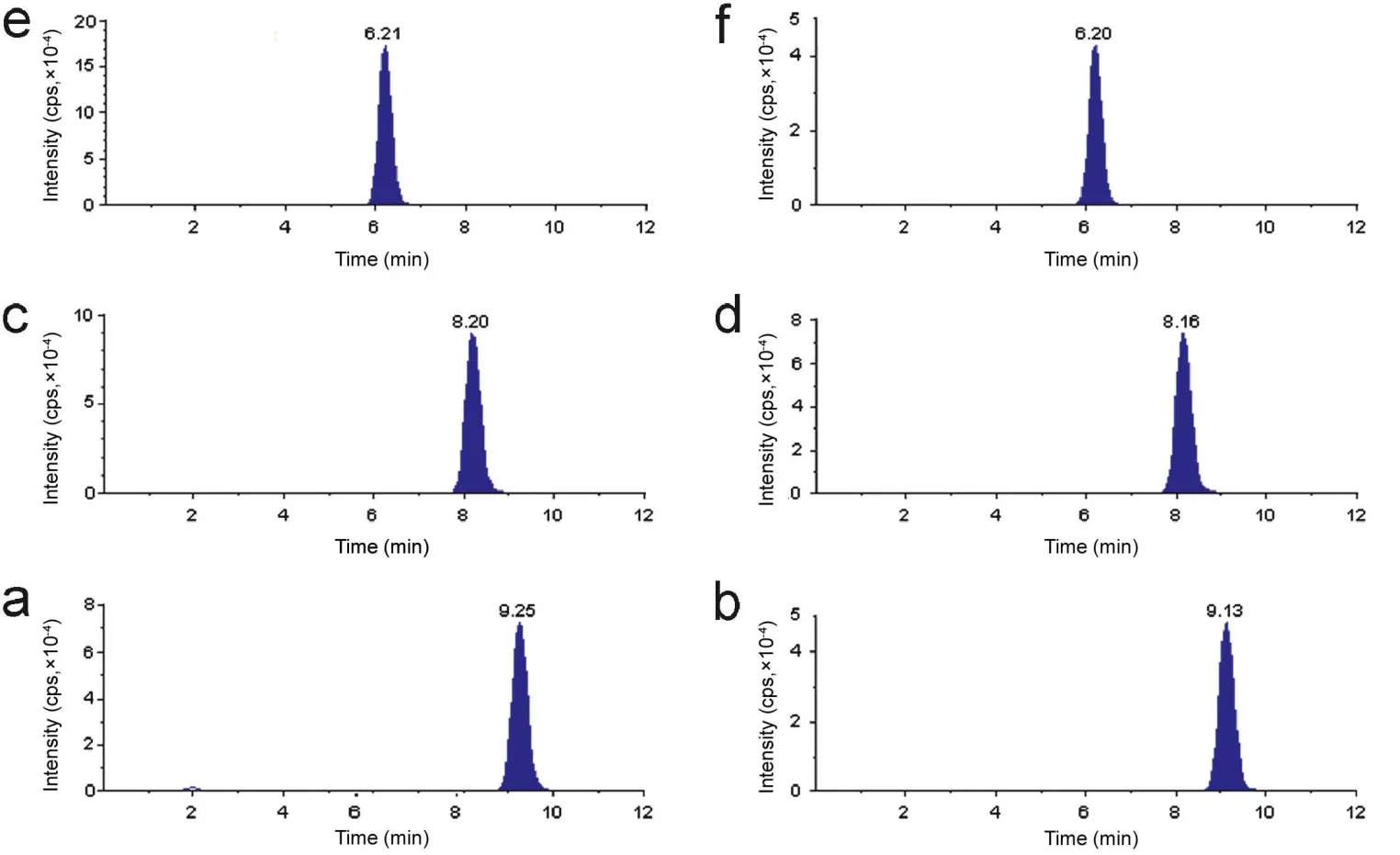
Figure 3 Chromatogram of(a)sibutramine,(b)sibutramine d7,(c)N-des methyl sibutramine,(d)N-des methyl sibutramine d7,(e)N-di des methyl sibutramine and (f) N-di des methyl sibutramine d7.
3.4. Precision and accuracy
Precision and accuracy of this method were evaluated by calculating within-run and between-run variations at three concentrations(30.0,3500.0 and 8000.0 pg/mL)of QC samples in six replicates. As shown in Table 3, within-run precision and accuracy were in the range of 1.3-2.9% and 99.0-99.9%for SB, 1.6-3.4% and 97.1-98.7% for DSB, 1.6-2.8 and 96.3-98.7% for DDSB, respectively. Similarly, between-run precision and accuracy were in the range of 1.6-2.8% and 99.4-101.7% for SB, 1.2-3.2% and 99.3-99.8% for DSB, 2.1-3.4%and 98.0-100.4%for DDSB,respectively.These results indicate that the method showed adequate reliability and reproducibility within the calibration curve range.
3.5. Matrix effect
The ion suppression/enhancement in the signal at MQC level was found to be CV 1.27 for SB, 1.20 for DSB and 1.30 for DDSB. These results indicate that there is minimum effect on ion suppression and ion enhancement at analyte and IS retention time.
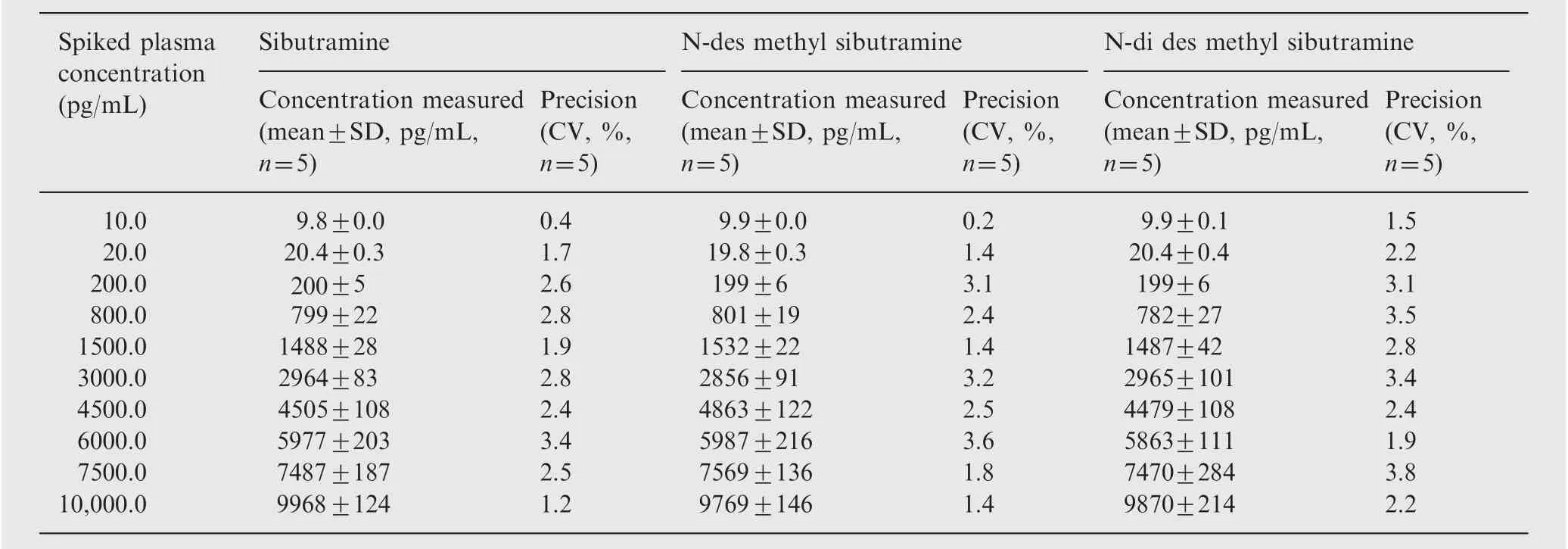
Table 2 Calibration curve details of sibutramine, N-des methyl sibutramine, N-di des methyl sibutramine.

Figure 4 Blank plasma chromatograms of sibutramine, sibutramine d7, N-des methyl sibutramine, N-des methyl sibutramine d7, N-di des methyl sibutramine and N-di des methyl sibutramine d7.
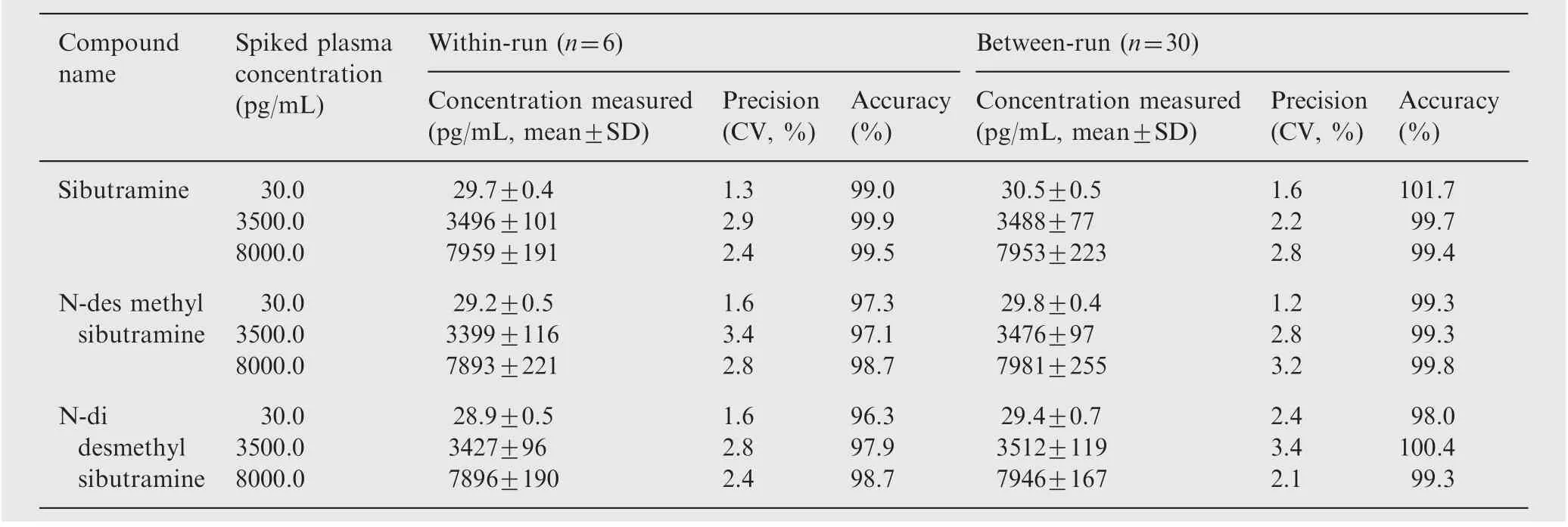
Table 3 Precision and accuracy (analysis with spiked plasma samples at three different concentrations).
3.6. Recovery
The extraction recoveries of SB, DSB and DDSB were determined at three different concentrations 30.0, 3500.0,8000.0 pg/mL and found to be 99.6±3.5%, 88.2±2.7% and 97.60±4.7%for SB,95.5±9.7%,92.6±10.2%and 92.3±4.7%for DSB, 99.2±2.4%, 94.2±1.7% and 94.5±6.1% for DDSB, respectively. The overall average recoveries of SB,DSB, DDSB and SB d7, DSB d7, DDSB d7 were found to be 95.1±6.1%, 93.5±1.8%, 96.0±2.8% and 98.1±4.5%,98.3±7.1%, 98.7±6.4%, respectively. Recoveries of the analyte and IS were consistent, precise and reproducible.

Table 4 Stability of sibutramine, N-des methyl sibutramine, N-di des methyl sibutramine in human plasma samples.
3.7. LOD and LOQ
The limit of detection was used to determine the instrument detection levels for SB, DSB, DDSB at low concentrations.5 μL of 0.5 pg/mL SB, DSB, DDSB solution was injected and the determined LOD was 4.5 fg for SB, 3.5 fg for DSB and 4.0 fg for DDSB with S/N values ≥3-5. The signal-to-noise(S/N) values found for six injections of SB, DSB, and DDSB at LOQ concentration were 31.95, 40.23 and 44.12,respectively.
3.8. Stability
Stock solution stability study was performed to check stability of SB, DSB, DDSB and SB d7, DSB d7, DDSB d7 stock solutions at 2-8°C in a refrigerator. The freshly prepared stock solutions were compared with stock solutions prepared 26 days ago. The changes of SB, DSB, DDSB and SBd7,DSBd7, DDSBd7 were -0.02% and 0.03% respectively,indicating that the stock solutions were stable for at least 26 days. Room temperature stability, autosampler stability,freeze-thaw stability and long term stability for SB, DSB and DDSB were investigated at LQC and HQC levels. The results revealed that SB, DSB and DDSB were stable in plasma for at least 72 h at room temperature, and 78 h in an autosampler. Freezing and thawing (three cycles) of plasma samples spiked with SB, DSB and DDSB at LQC and HQC levels did not affect their stability. The long-term stability results also indicated that SB, DSB and DDSB were stable in plasma up to 71 days at 30°C. The results obtained from all these stability studies are tabulated in Table 4. Precision (CV)is less than 5% for room temperature, long-term, Freezethaw, and autosampler stabilities.
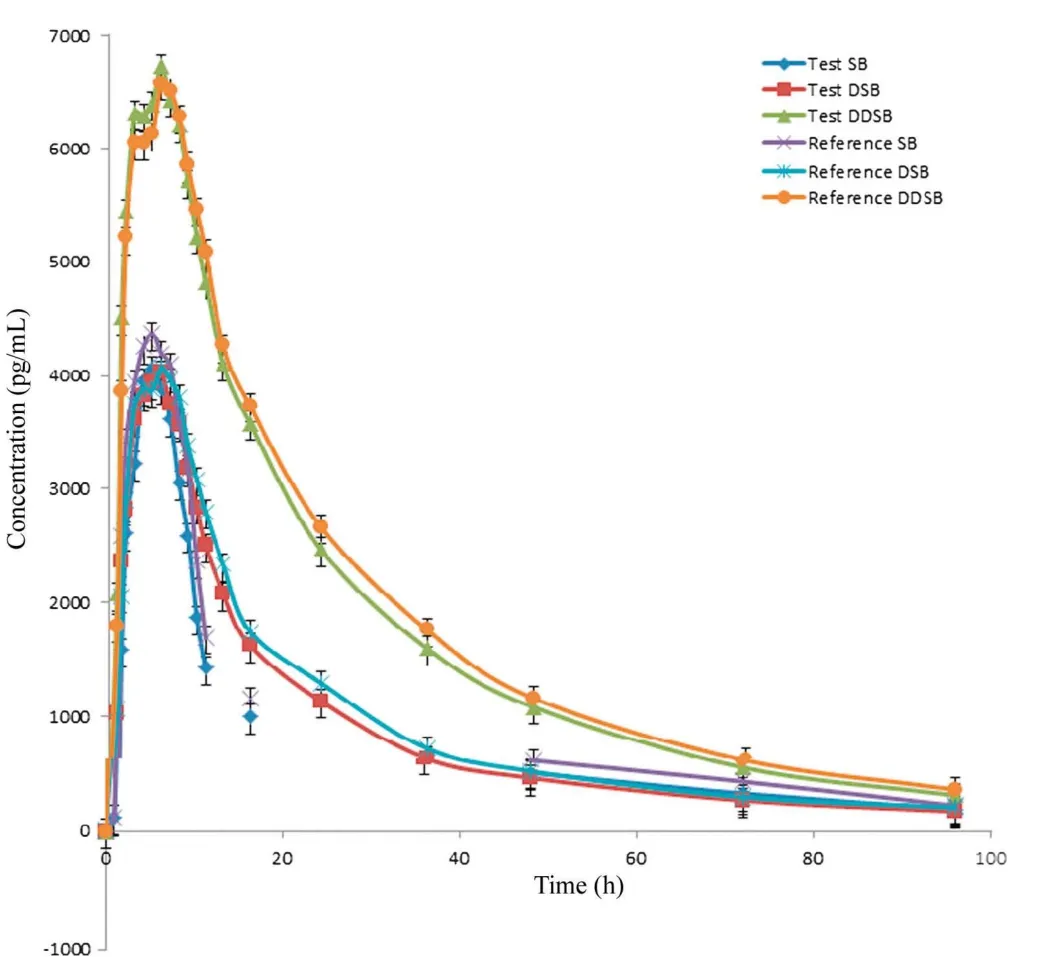
Figure 5 Mean plasma concentration of sibutramine, N-des methyl sibutramine, and N-di des methyl sibutramine.
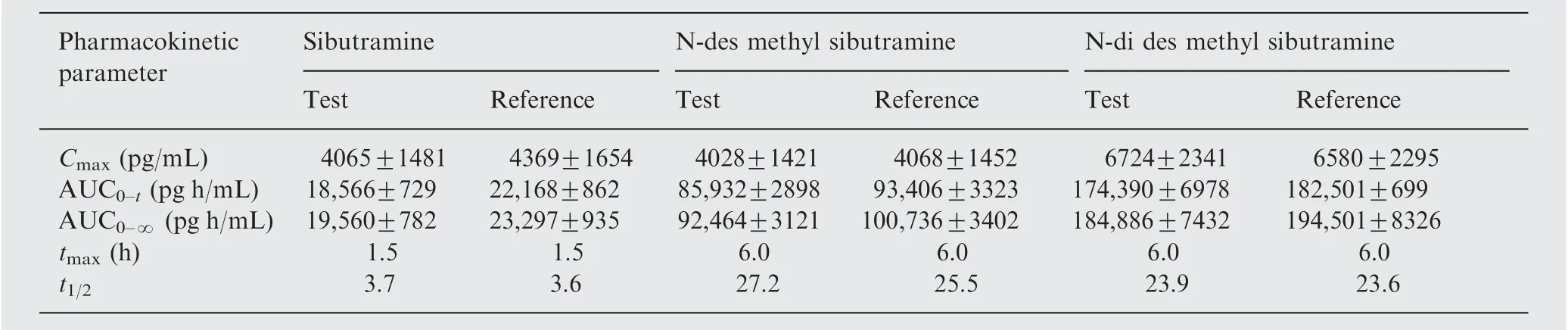
Table 5 Mean pharmacokinetic parameters of sibutramine, N-des methyl sibutramine, and N-di des methyl sibutramine in 40 healthy human volunteers after oral administration of 15 mg test and reference products (mean±SD).

Table 6 Test vs.reference pharmacokinetic parameters of sibutramine, N-des methyl sibutramine and N-di des methyl sibutramine after administration of 15 mg of test and reference products in 40 healthy human volunteers.
3.9. Application to biological samples
The validated method was used successfully to quantify plasma SB, DSB and DDSB concentrations of 40 healthy volunteers who received oral administration of 15 mg dose(one 15 mg capsule) under fasting conditions. Mean plasma concentration versus time profiles are shown in Fig.5. All the plasma concentrations of SB,DSB and DDSB were within the standard calibration region and remained above LOQ(10.0 pg/mL) for the entire sampling period. The detailed pharmacokinetic parameters are shown in Tables 5 and 6. In addition, the mean ratio of AUC0-t/AUC0-∞was higher than 90% following the FDA Bioequivalence Guidelines [26,27].The ratio of test/reference(T/R)for overall analysis was in the range of 80-125%.
4. Conclusion
The developed method is highly specific due to the inherent selectivity of tandem mass spectrometry and has significant advantages over previously reported methods in terms of selectivity, sensitivity, linearity and reproducibility. Quantification of sibutramine and its two metabolites was compared with respective isotope labeled internal standards. Extraction of analyte and IS was achieved using liquid-liquid extraction.Mobile phase, column, flow rate, injection volume, plasma usage volume for analysis were improved. The bioequivalence study also demonstrated that the two sibutramine formulations (reference and test) are bioequivalent.
Authors wish to thank the support received from IICT (Indian Institute of Chemical Technology), Hyderabad, India for providing literature survey, APL Research Pvt. Ltd., Hyderabad, India to carry out this Research work.
[1] E.Kristina,R.Stephan,B.Britta,et al.,Sibutramine treatment in obesity: initial eating behaviour in relation to weight loss results and changes in mood, Pharmacol. Res. 51 (2005) 159-163.
[2] K. Heusser,J. Tank,A.Diedrich, et al.,Influence of sibutramine treatment on sympathetic vasomotor tone in obese subjects,Clin.Pharmacol. Ther. 79 (2006) 500-508.
[3] D. Sucar, E.B. Sougey, J.B. Neto, Psychotic episode induced by potential drug interaction of sibutramine and finasteride, Rev.Bras. Psiquiatr. 24 (2002) 30-33.
[4] Y.Chen,L.Zhao,F.Lu,et al.,Determination of synthetic drugs used to adulterate botanical dietary supplements using QTRAP LC-MS/MS, Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 26 (5) (2009) 595-603.
[5] P.Zou,S.S.Oh, K.H.Kiang,et al.,Detection of sibutramine,its two metabolites and one analogue in a herbal product for weight loss by liquid chromatography triple quadrupole mass spectrometry and time-of-flight mass spectrometry, Rapid Commun.Mass Spectrom. 21 (4) (2007) 614-618.
[6] J. Chen, W. Lu, Q. Zhang, et al., Determination of the active metabolite of sibutramine by liquid chromatography-electrospray ionization tandem mass spectrometry, J. Chromatogr. B Anal.Technol. Biomed. Life Sci. 785 (2) (2003) 197-203.
[7] S.H.Kim,J. Lee, T.Yoon,et al.,Simultaneous determination of anti-diabetes/anti-obesity drugs by LC/PDA, and targeted analysis of sibutramine analog in dietary supplements by LC/MS/MS, Biomed. Chromatogr. 12 (2009) 1259-1265.
[8] I.C. Diefenbach, M. Friedrich, M.R. Dos, et al., Development and validation of a column high-performance liquid chromatographic method for determination of sibutramine in capsules,J. AOAC Int. 92 (1) (2009) 148-151.
[9] Z.Huang,S.Xiao,D.Luo,et al.,Simultaneous determination of sibutramine and N-Di-desmethylsibutramine in dietary supplements for weight control by HPLC-ESI-MS,J. Chromatogr.Sci.46 (8) (2008) 707-711.
[10] A.K. Singh, L.G. Pedro, F.P. Gomes, et al., Development and validation of sensitive methods for determination of sibutramine hydrochloride monohydrate and direct enantiomeric separation on a protein-based chiral stationary phase, J. AOAC Int. 91 (3)(2008) 572-579.
[11] J.G. Chandorkar, V.B. Kotwal, N.S. Dhande, et al., Development and validation of high performance liquid chromatography method for analysis of sibutramine hydrochloride and its impurity, Pak. J. Pharm. Sci. 21 (2) (2008) 121-124.
[12] K.S. Hakala, M. Link, B. Szotakova, et al., Characterization of metabolites of sibutramine in primary cultures of rat hepatocytes by liquid chromatography-ion trap mass spectrometry, Anal.Bioanal. Chem. 393 (4) (2009) 1327-1336.
[13] S.Y.Um,K.B.Kim,S.H.Kim,et al.,Determination of the active metabolites of sibutramine in rat serum using column-switching HPLC, J. Sep. Sci. 31 (15) (2008) 2820-2826.
[14] A. Balcioglu, R.J. Wurtman, Sibutramine, a serotonin uptake inhibitor, increases dopamine concentrations in rat striatal and hypothalamic extracellular fluid, Neuropharmacology 39 (12)(2000) 2352-2359.
[15] D.J. Heal, A.T. Frankland, W.R. Buckett, et al., A new and highly sensitive method for measuring 3-methoxytyramine using HPLC with electrochemical detection. Studies with drugs which alter dopamine metabolism in the brain, Neuropharmacology 29(12) (1990) 1141-1150.
[16] D.J. Heal, M.R. Prow, W.R. Buckett, et al., Measurement of 3-methoxy-4-hydroxyphenylglycol (MHPG) in mouse brain by HPLC with electrochemical detection, as an index of noradrenaline utilisation and presynaptic alpha 2-adrenoceptor function,Br. J. Pharmacol. 96 (3) (1989) 547-556.
[17] A.C. Franco Spinola, S. Almeida, A. Filipe, et al., Comparative bioavailability of two formulations of sibutramine, Int. J. Clin.Pharmacol. Ther. 47 (10) (2009) 627-637.
[18] W. Kang, K. Bae, K. Noh, et al., Enantioselective determination of sibutramine and its active metabolites in human plasma,J. Pharm. Biomed. Anal. 51 (1) (2010) 264-267.
[19] D.S. Jain, G. Subbaiah, M. Sanyal, et al., Liquid chromatography/electrospray ionization tandem mass spectrometry validated method for the simultaneous quantification of sibutramine and its primary and secondary amine metabolites in human plasma and its application to a bioequivalence study, Rapid Commun. Mass Spectrom. 20 (23) (2006) 3509-3521.
[20] Z. Abolfathi, J. Couture, F. Vallee, et al., A pilot study to evaluate the pharmacokinetics of sibutramine in healthy subjects under fasting and fed conditions, J. Pharm. Pharm. Sci. 7 (3)(2004) 345-349.
[21] W. Kang, K. Bae, K. Noh, et al., Enantioselective determination of sibutramine and its active metabolites in human plasma,J. Pharm. Biomed. Anal. 51 (1) (2010) 264-267.
[22] L.Ding,X.Hao,X.Huang,et al.,Simultaneous determination of sibutramine and its N-desmethyl metabolites in human plasma by liquid chromatography-electrospray ionization-mass spectrometry: method and clinical applications, Anal. Chim. Acta 492(2003) 241-248.
[23] J. Chen, W. Lu, Q. Zhang, et al., Determination of the active metabolite of sibutramine by liquid chromatography-electrospray ionization tandem mass spectrometry, J. Chromatogr. B 785(2003) 197-203.
[24] J.W. Bae, C.I. Choi, C.G. Jang, et al., Simultaneous determination of sibutramine and its active metabolites in human plasma by LC-MS/MS and its application to a pharmacokinetic study,Biomed. Chromatogr. (2011), doi:10.1002/bmc.1587.
[25] Guidance for Industry: Bioanalytical Method Validation, U.S.Department of Health and Human Services, Food and Drug Administration,Center for Drug Evaluation and Research(CDER),Center for Biologics Evaluation and Research (CBER), May 2001.
[26] Guidance for Industry: Food-effect Bioavailability and Fed Bioequivalence Studies, U.S. Department of Health and Human Services, Food and Drug Administration, Centre for Drug Evaluation and Research (CDER), December 2002.
[27] Guidance for Industry: Bioavailability and Fed Bioequivalence Studies for Orally Administered Drug Products—General Considerations, U.S. Department of Health and Human Services,Food and Drug Administration,Centre for Drug Evaluation and Research (CDER), March 2003.
 Journal of Pharmaceutical Analysis2012年4期
Journal of Pharmaceutical Analysis2012年4期
- Journal of Pharmaceutical Analysis的其它文章
- Two spectrophotometric methods for simultaneous determination of some antihyperlipidemic drugs
- Azeotropic mixture used for development and validation of Lornoxicam in bulk and its tablet dosage form by spectrophotometric method
- Application of analytical instruments in pharmaceutical analysis
- Rapid analysis of piperazine ferulate tablets by optic-fiber sensing technology and the similarity of ultraviolet spectra
- Schisandra chinensis (Turcz.) Baill.
- Stability-indicating liquid chromatographic method for the determination of Letrozole in pharmaceutical formulations