还原剂对多孔阳极氧化铝纳米孔道结构的影响
2012-12-11 09:24朱绪飞马宏图戚卫星
物理化学学报 2012年11期
朱绪飞 韩 华 马宏图 路 超 戚卫星 徐 晨
(1南京理工大学,软化学与功能材料教育部重点实验室,南京210094; 2中国科学院自动化研究所,国家专用集成电路设计工程技术研究中心,北京100190)
还原剂对多孔阳极氧化铝纳米孔道结构的影响
朱绪飞1,*韩 华2,*马宏图2路 超1戚卫星1徐 晨1
(1南京理工大学,软化学与功能材料教育部重点实验室,南京210094;2中国科学院自动化研究所,国家专用集成电路设计工程技术研究中心,北京100190)
多孔阳极氧化铝(PAA)和多孔阳极氧化钛纳米管(PATNT)的结构调控近年来倍受关注.在形成机理尚不清楚的情况下,对PAA和PATNT的结构调控很难避免盲目性.为验证“氧气气泡模具”可以形成纳米孔道这个新观点,本文采用化学方法对PAA的结构进行调控,成功地引入了一种还原剂来吸收纳米孔道中的氧气气泡.在添加还原剂的草酸溶液中得到了一种特殊的阳极氧化铝膜.研究了还原剂的含量对磷酸溶液中形成PAA孔道结构的影响,结果表明随着还原剂含量的增加,PAA的孔道直径逐渐减小,有序性降低.对比了添加还原剂前后阳极氧化过程的电压-时间曲线的差异,结果表明,在含有还原剂溶液中制备的阳极氧化铝膜的导电性明显提高.在密封的条件下,还原剂能吸收掉孔道中的氧气,使气泡模具效应消失,得到完全的致密型氧化膜.这些实验事实充分证明PAA中有序孔道的形成是氧气气泡模具效应的结果.
纳米多孔材料;阳极氧化;电解液;还原剂;形成机理
1 引言
通过阳极氧化法制备的多孔阳极氧化铝(PAA)和多孔阳极氧化钛纳米管(PATNT)由于其独特有序的纳米孔道结构和潜在应用价值而倍受关注.其中PAA作为组装各种纳米功能材料的首选模板至今仍被广泛采用;1-6PATNT作为一种无机光敏半导体材料,在光电催化、染料敏化太阳能电池、光解水等领域都有潜在应用价值,因此PATNT也成为近年来的热门课题.7-9纵观PAA和PATNT的研究内容主要包括形成机理、结构调控及其应用等三个方面.相比而言,结构调控和制备的新技术和新工艺研究更受关注.10-13PNTNT结构调控的新技术一般来源于PAA制备技术,例如2006年Lee等14发表在Nature Materials上的论文报道了采用硬氧化和温和氧化交替的新工艺,制备了孔道直径呈周期变化的长程有序PAA模板;2008年他们又在Nature Nanotechnology期刊上报道采用脉冲阳极氧化技术制备了孔径周期性变化和大面积分层的PAA模板,15为后来PAA模板的制备与调控起了导向作用.随后各国研究者因组装各种纳米功能材料的需要,通过改变阳极氧化条件制备了各种特殊的(分叉形、锯齿形、骨形等)PAA模板,叶秋梅等16已经对这些特殊模板的制备技术进行了综述.这些制备技术也被借鉴到PATNT结构的调控中,例如,北京大学的张耿民等17改进了传统的阳极氧化条件,创新性地采用直流与交流电压交替使用,制备了光滑管壁和竹子结构的多段PATNT;上海交通大学的陈险峰等18采用脉冲氧化技术制备了结构周期性变化的PATNT;中国科学院兰州化学物理研究所的Wang等9在氟化铵和氢氟酸的乙二醇有机溶液中,制备了长程有序的PATNT,解决了常规氧化中纳米管的集束打捆问题.总之,PAA和PATNT的制备与结构调控至今仍是被广泛关注的领域,研究论文层出不穷.19-25
与制备方面诸多高档次文章相比,对形成机理的研究相对较少且难度较大,因为这涉及到众多学科(化学、物理化学、金属腐蚀与防护、凝聚态物理、物理电子学等)的基础理论问题,尽管PAA形成机理已经研究了几十年,但至今纳米孔道的产生机理和自组织过程仍无统一的认识.26,27但要实现制备与应用技术的原始创新,对PAA和PATNT形成机理的研究就显得非常必要和迫切,仅2012年最新出版的机理研究文章有四篇.28-31作者的团队有幸得到国家基金的资助对多孔氧化物形成机理进行专门研究,最近我们对比综述了PAA和PATNT形成机理的最新进展,32并提出了氧气气泡模具效应成孔的新观点.铝和钛阳极氧化过程中微量氧气的析出及其对纳米孔道形成的影响已经被最新的机理研究文献关注和引用.28-31
纵观PAA和PATNT结构调控一般是通过物理方法(改变电流和电压的施加方式)来实现的,其实电解液组分对PAA形貌也有很大影响.北京大学的吴凯等33曾研究了贫水电解液中PAA模板的生长,认为电解液中水分含量的降低能有效地抑制电化学氧化和溶解速率,在乙二醇等有机电解液中(与水溶液相比)能得到更加有序的PAA模板,且施加的氧化电压范围更宽.天津大学的阴育新等34研究了电解液中的阴离子对PATNT生长的影响,结果表明PATNT的生长速率和长度依赖于阴离子的种类与浓度.本文基于我们提出的氧气气泡模具成孔的观点,32,35,36从改变电解液组分、添加还原剂吸收纳米孔道中的氧气来改变PAA孔道的形貌结构,采用这种化学的方法来实现PAA的结构调控尚未见报道.众所周知,PATNT应用难点是如何降低PATNT的禁带宽度、提高其光电转换效率,这种既能改变孔道结构又能改变纳米管中阴离子组成的化学调控方法,37,38对解决PATNT这一难点有重要的借鉴意义,研究者可以从各自的学科背景出发,提出更多的原始创新的思想和方法.
2 实验
本文采用的是铝电解电容器专用的高纯度铝箔(纯度99.99%,厚度0.2 mm,南京春田电子有限公司)和钛箔(纯度99.5%,厚度0.1 mm,宝鸡荣豪钛业公司),其他化学试剂的纯度均为分析纯.实验用铝片(10 mm宽,80 mm长)的前处理工艺是:用载玻片将铝片表面压平整,而后放入2%(w)氢氧化钠水溶液中于75°C下浸泡2 min,除去铝片表面的天然氧化膜,而后用去离子水冲洗干净,吹干后马上进行电化学抛光.将铝片浸入75°C的抛光液(磷酸:三氧化铬:水质量比为80:12:8)中,以120 mA·cm-2的电流密度进行恒电流电化学抛光90 s,使铝片表面成为光亮的镜面,马上用去离子水洗净抛光后的铝片,晾干待用.实验用钛片(10 mm宽,80 mm长)的前处理工艺是:先用丙酮将钛片表面的油脂洗净,用载玻片将钛片表面压平整,用去离子水冲洗干净,吹干后的钛片马上进行化学抛光60 s.抛光液是由氢氟酸与硝酸按体积比1:3混合而成,取出抛光后的钛片马上用去离子水洗净,晾干待用.
钛的阳极氧化是在1%(w)氟化铵的乙二醇溶液中进行的.铝的阳极氧化采用了2%(w)草酸溶液和2%(w)磷酸溶液为基础电解液.因为在单纯的磷酸水溶液中在常温下阳极氧化时会出现分叉形、锯齿形等不规则形状的孔道,因此本文统一选用乙二醇和去离子水按质量比1:1组成的混合溶剂配制了相应的草酸和磷酸的溶液,而后分别再添加不同含量的还原剂次亚磷酸铵(NH4H2PO2)、水合肼(N2H4· H2O)和非还原性的碳酸氢铵、己二酸铵得到相应的实验电解液.
阳极氧化过程分别在相应的电解液中在室温水浴条件下进行,采用恒定电流的阳极氧化方式,电压-时间曲线的测定是采用南京理工大学自行研制的SY-98型阳极氧化计算机测试系统,该系统数据采集时间短(只有10 ms),这样能确保采集到每一个微小的电压波动信号,数据处理也由计算机自动完成.阳极氧化结束后的样品,在去离子水中浸泡20 min后自然晾干待扫描电镜表征.本文使用日本Hitachi-S4800 II型、德国LEO-1530VP和Zeiss SUPRA 55场发射扫描电镜(SEM)对样品形貌进行表征.
3 结果与讨论
3.1 常规PATNT和PAA的纳米孔道结构
图1是在恒定电流密度(10 mA·cm-2)下阳极氧化得到的PATNT和PAA孔道断面形貌的SEM照片和相应的电压-时间曲线.图1a是钛在1%(w)氟化铵溶液中阳极氧化得到的PATNT的断面形貌,每根纳米管之间的缝隙清晰可见,也没有看到纳米管外围“腰带式”的凸纹.26-28通常认为PATNT中“腰带式”凸纹的出现是不可避免的,最新出版的文献28提出“腰带式”凸纹是围绕着纳米管周围生长的,随着阳极氧化时间的延长,最靠近表面层的“腰带式”凸纹会被溶解掉,变成光滑的PATNT;靠近底部的“腰带式”凸纹没有被溶解就会依然存在.28图1a是在恒定电流密度(10 mA·cm-2)下阳极氧化400 s得到的PATNT,在靠近表面层的纳米管也没有看到这种“腰带式”的凸纹.可见,关于PATNT的形成与溶解机制还存在争议.Schmuki等26认为氟离子穿过PATNT底部的阻挡层,在金属界面的富集是形成纳米管之间缝隙的主要原因;其实在PAA的形成过程中根本没有氟离子存在,但也能形成一个个独立的元胞结构.14-16Su和Zhou27认为PATNT之间缝隙的形成尚无法解释.最近我们在最新的综述32中提出了氧气气泡模具形成规则孔道的观点,无论是PAA六棱柱独立元胞之间的缝隙,还是PATNT之间的缝隙,都是由于底部阻挡层附近“氧气气泡模具效应”导致了半球形孔洞胚胎的形成,气泡冒出后电解液进入孔道底部后,呈半球形分布的电场导致孔壁氧化物围绕氧气气泡模具“从下向上”生长的结果.这符合Skeldon等39,40提出的PAA孔壁是由于阻挡层氧化物向两侧流动迁移的观点.但是由于纳米孔道中析出的氧气极其微少(按照库仑定律,在20 mA·cm-2电流密度下阳极氧化,假设其中电子电流是2 mA· cm-2时,阳极氧化3600 s时间内析出氧气的量是0.597 mg),同时纳米尺度的氧气气泡难免有部分要溶解到电解液中,因此在水溶液中几乎无法看到氧气气泡的冒出,在粘度较大的乙二醇或甘油溶液中,或者真空条件下或大电流密度下能看到氧气的析出.41-43正如Patermarakis和Moussoutzanis44指出:由于PAA的阳极氧化过程是在液体中施加外加电源的情况下进行的,无法用原位表征技术进行动态跟踪,因此要得到纳米孔道中氧气气泡的直接证据是相当困难的.
为了验证氧气气泡成孔的观点,本文采用在电解液中添加还原剂的化学方法来进行间接的验证.图1a是在1%(w)NH4F溶液中得到的PATNT的断面形貌,图1b是相应的电压-时间曲线.可见,在PATNT中只能看到整个纳米管断层的形貌,在我们已阅读的文献中尚没看到纳米管内部的剖面结构,也就是说,即使PATNT内部结构形状发生了变化,也很难从SEM照片中直接观察到.但是,PAA纳米孔道内部的剖面结构却很容易从PAA膜的裂缝中观察到,图1c和图1e分别是在2%(w)磷酸和2% (w)草酸溶液中得到PAA孔道形貌结构的照片.图1d和图1f是它们对应的阳极氧化过程的电压-时间曲线.从三条电压-时间曲线可见,PATNT和PAA的形成过程和形成机制是相似的,26-30我们也认同这个观点,唯一的差别在于氧化铝和氧化钛力学性能或者膨胀系数的不同,这也是造成PAA和PATNT之间缝隙宽度不同和断裂形式差异的本质原因.32因此,本文侧重研究在形成PAA的磷酸和草酸溶液中,添加还原性和非还原性电解质前后对PAA孔道内部结构的影响.
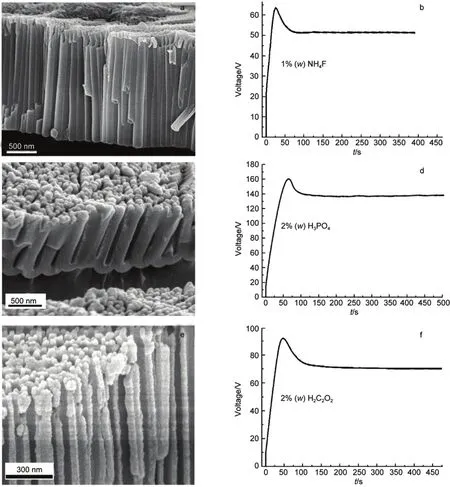
图1 多孔阳极氧化钛纳米管(PATNT)的断面形貌(a)及其电压-时间曲线(b);在H3PO4溶液中得到的多孔阳极氧化铝(PAA)的形貌(c)及其电压-时间曲线(d);在H2C2O4溶液中得到的PAA的形貌(e)及其电压-时间曲线(f)Fig.1 Morphology of the cross-section of porous anodic TiO2nanotubes(PATNT)(a)and the voltage-time curve(b); morphology of porous anodic alumina(PAA)anodized in H3PO4solution(c)and the voltage-time curve(d);morphology of PAAanodized in H2C2O4solution(e)and the voltage-time curve(f)
3.2 还原剂对草酸溶液中PAA孔道结构的影响
图2是在2%(w)草酸溶液中添加还原剂(NH4H2PO2)前后所得到PAA形貌结构的对比照片.图2a和图2b是在2%(w)H2C2O4溶液中恒定电流密度(10 mA·cm-2)下阳极氧化500 s得到的PAA膜的SEM照片.图2c、图2d和图2e是在2%(w)H2C2O4+ 8%(w)NH4H2PO2溶液中阳极氧化得到的阳极氧化铝膜的SEM照片,其它阳极氧化条件与图2a相同.从图2c和图2d的形貌照片可见,添加还原剂以后,这种特殊的阳极氧化铝膜发生了很大变化,既不是传统的致密型的阳极氧化铝,也不是多孔阳极氧化铝,31-33本文称之为特殊的阳极氧化铝膜.图2a和图2c的表面形貌照片的放大倍数是相同的,但添加还原剂后的特殊阳极氧化铝膜表面只能看到无数杂乱无章的微小孔,从图2e更大放大倍数的照片可见,这些不规则的孔道时断时续,与添加还原剂前的图2b相比其孔道结构已发生明显的变化.我们认为这种形貌结构的巨大差异是还原剂吸收了纳米孔道底部析出的氧气气泡造成的,随着氧气气泡模具效应减弱或消失,因此规则的半球形(与图2b相比较)结构也随之消失.图3是铝在两种电解液中阳极氧化时的电压-时间曲线对比图.尽管添加还原剂后电压-时间曲线的形状仍然是PAA形成曲线的形状,30,32但水平电压降低了大约20 V,抛开电解液电导率引起的压降差(只有5 V左右),45说明这种特殊阳极氧化铝膜的导电性优于传统的PAA膜,这给我们一个启示,在添加还原剂的电解液中形成的特殊阳极氧化铝膜,不但形貌结构发生很大变化,同时也告诉我们特殊氧化膜的导电性要优于传统PAA膜阻挡层的导电性,这一点对于改善PATNT的导电性或者禁带宽度将有重要的借鉴意义.

图2 在2%(w)H2C2O4溶液中得到的PAA的表面形貌(a)和断面形貌(b);在2%(w)H2C2O4+8%(w)NH4H2PO2溶液中得到的阳极氧化铝的表面形貌(c)和断面形貌(d,e)Fig.2 Morphologies of the surface(a)and the cross-section(b)of PAAanodized in 2%(w)H2C2O4solution;morphologies of the surface(c)and the cross-section(d,e)of the anodic alumina anodized in 2%(w)H2C2O4+8%(w)NH4H2PO2solution
3.3 还原剂对磷酸溶液中PAA孔道结构的影响
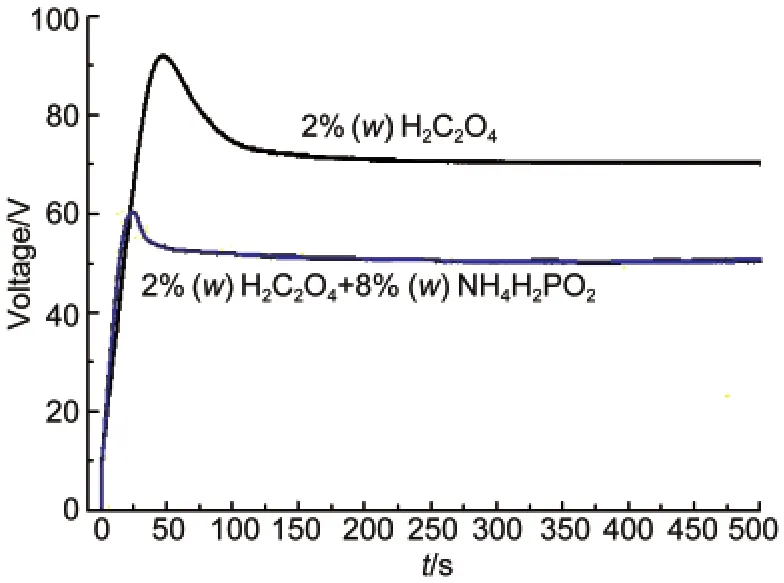
图3 铝在两种电解液中阳极氧化时的电压-时间曲线Fig.3 Voltage-time curves of the aluminium anodizing in two kinds of electrolytes
为了进一步验证“氧气气泡模具效应”成孔的观点,本文在2%(w)磷酸溶液的基础上分别添加不同含量的还原剂,在其它阳极氧化条件都一样的情况下,得到了各种形态结构的PAA孔道.图4是不同含量还原剂对磷酸电解液中PAA孔道结构的影响.图4a、图4b和图4c分别添加还原剂的比例是2%、4%和6%(w).与图1c规则有序的PAA孔道相比,图4中的各种PAA孔道在结构上发生了巨大的变化,随着还原剂含量的增加,孔道直径与图1c相比明显变小,例如图1c中孔道直径大约在100 nm以上,而图4a中孔道直径明显小于100 nm,图4b中的孔道直径又明显小于图4a,且更加无序.随着还原剂含量的进一步增加,图4d的表面已经看不见孔洞,图4c中间部位能明显看到几个上端封闭的极小孔道.与图1c和图2b孔道底部规则的半球形形状相比,图4中孔道底部形状已经变得不规则.由于还原剂在不同阶段可能吸收氧气气泡的效果不同,因此孔道上部和下部的形貌差别也很大,例如图4b中孔道底部有更多的小而短的孔道,说明在这时孔道底部氧气气泡被吸收的更多.总之,加入还原剂之后孔道形貌大大改变,自组织有序性大大降低.这些证据充分说明电解液中添加还原剂后PAA形貌结构的变化是由于还原剂对纳米孔道中氧气的吸收所造成,这从侧面充分证明了PAA中规则有序的纳米孔道是氧气气泡模具效应的结果.32,35从这些规则孔道消失的反面证据可见,PAA有序纳米结构的自组织过程也与氧气气泡的析出有必然的联系,10,32这对于清楚认识PAA和PATNT有序纳米结构的自组织本质也具有指导意义.

图4 在2%(w)H3PO4+2%(w)NH4H2PO2溶液(a);2%(w)H3PO4+4%(w)NH4H2PO2溶液(b);2%(w)H3PO4+ 6%(w)NH4H2PO2溶液(c,d)中氧化得到的PAA的断面形貌Fig.4 Morphologies of the cross-section of PAAanodized in 2%(w)H3PO4+2%(w)NH4H2PO2solution(a), 2%(w)H3PO4+4%(w)NH4H2PO2solution(b);and 2%(w)H3PO4+6%(w)NH4H2PO2solution(c,d)
图5给出了铝在2%(w)H3PO4+4%(w) NH4H2PO2溶液中阳极氧化时的电压-时间曲线.与单纯的2%(w)H3PO4溶液中阳极氧化时的电压-时间曲线(图1d)相比有两点明显的差异:第一是在添加还原剂后曲线有明显上升趋势,这说明多孔型氧化膜有向致密型氧化膜转换的趋势;32第二是水平电压下降60 V左右,这也说明图4中的阳极氧化膜的导电性要比图1c中PAA膜或者阻挡层的导电性要更好一些,这与图3中的结论是一致的.
3.4 其它电解质添加后对PAA孔道结构的影响
前面讨论了还原剂次亚磷酸铵加入电解液后对PAA孔道形貌的影响,研究者也许会有这样的疑问,次亚磷酸铵的加入到底是改变了阳极氧化过程中的电化学氧化反应还是通过吸收O2气泡来改变了孔道形状?添加非还原性的电解质是否也能改变孔道形貌呢?为此,本文分别针对两种非还原性的电解质(碳酸氢铵和己二酸铵)和两种还原性电解质(水合肼和次亚磷酸铵)进行对比试验,并在3.5节中对电化学反应过程是否改变进行了讨论.
再以2%(w)H2C2O4溶液为基础电解液,分别添加4%(w)碳酸氢铵、4%(w)己二酸铵(N2H16C6O4)、4%(w)水合肼(N2H4·H2O)和4%(w)的次亚磷酸铵组成四种新的电解液,在这四种新的电解液中分别进行恒电流阳极氧化(10 mA·cm-2)500 s后,图6是在四种不同电解液得到的阳极氧化膜的SEM照片.与基础电解液中得到的PAA孔道的断面形貌(图2b)相比,图6(a,b)中的孔道结构并未发生形状上的改变,表面孔洞也清晰可见,这说明添加非还原性的碳酸氢铵和己二酸铵组分并没有影响到孔道的结构.而在草酸电解液中添加强还原剂水合肼以后,经过阳极氧化后直接得到了致密型的氧化膜(图6c),表面裂纹或者孔洞胚胎甚至也没有看到;形成这种致密型氧化膜的原因可能是水合肼直接抑制了氧气的产生,也可能是水合肼的强碱性中和了草酸的酸性.与图6c的致密膜相比,显然图2和图4中的多孔膜应该首先形成纳米孔道,而后还原剂次亚磷酸铵吸收了孔道底部的部分氧气,气泡模具形状的改变导致了图2和图4孔道结构的改变.否则,假若次亚磷酸铵与水合肼一样直接抑制了氧气的产生,图2和图4中的多孔膜就变成图6c中的致密膜了.
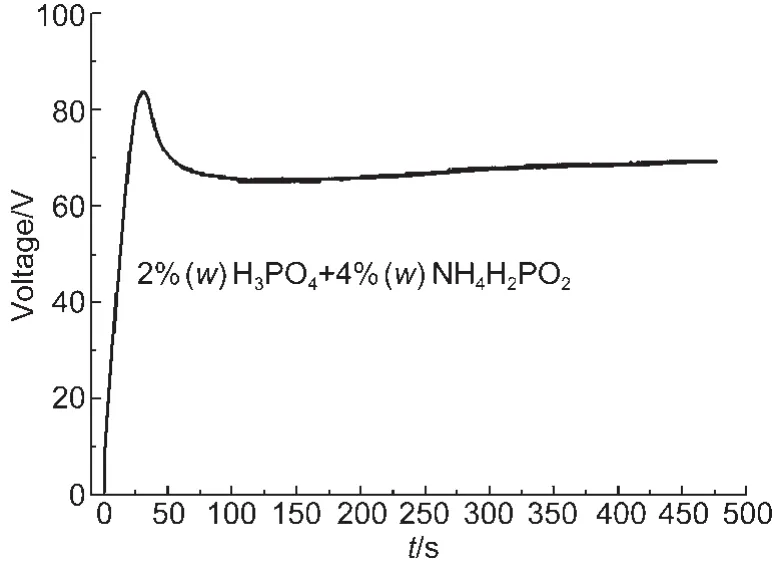
图5 铝在2%(w)H3PO4+4%(w)NH4H2PO2溶液中阳极氧化时的电压-时间曲线Fig.5 Voltage-time curve of the aluminium anodizing in 2%(w)H3PO4+4%(w)NH4H2PO2solution

图6 在四种不同添加剂的电解液中氧化后得到的阳极氧化铝膜的断面形貌Fig.6 Morphologies of the cross-sections of the anodic alumina films anodized in different electrolytes(a)NH4HCO3;(b)N2H16C6O4;(c)N2H4·H2O;(d)NH4H2PO2
图6d是铝在2%(w)草酸添加4%(w)次亚磷酸铵组成的混合电解液中阳极氧化后得到的氧化膜的形貌,与图2d相比其孔道有序性更高一些,这是由于还原剂含量降低引起的.可见,在磷酸、草酸等典型的PAA电解液中,添加不同含量的次亚磷酸铵,都能改变其阳极氧化膜的孔道结构.
3.5 PAA孔道结构改变的电化学反应机理
Li等30在最新文章中综述了铝阳极氧化过程的电化学反应.普遍认为:不管是在磷酸、草酸、硫酸等形成多孔型氧化膜的电解液中,还是在己二酸铵、碳酸氢铵、五硼酸铵等形成致密型氧化膜的电解液中,32主要的电化学反应包括三个过程:在初始阶段的水电解过程(I)、氧化铝的形成过程(II)和氧化铝被溶解的过程(III).30其中水电解过程(I)中可能的反应有5个,包括阳极反应(2)、(3)、(4)和阴极反应(5):
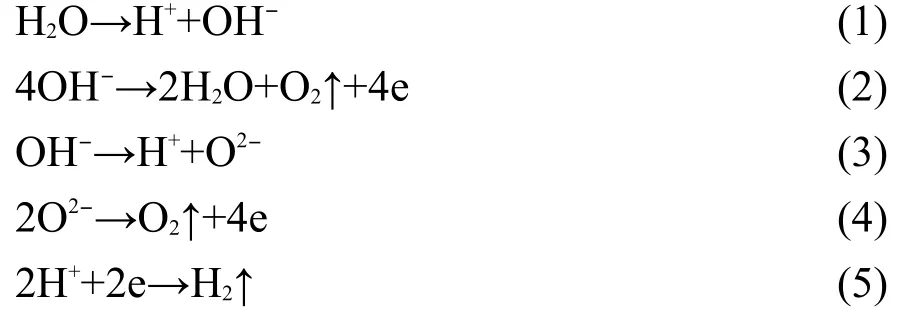
氧化铝在阳极的形成过程(II)包括:

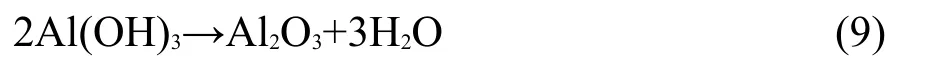
发生在阳极表面的Al2O3被溶解和孔洞的形成与加深过程(III)可能的反应有:
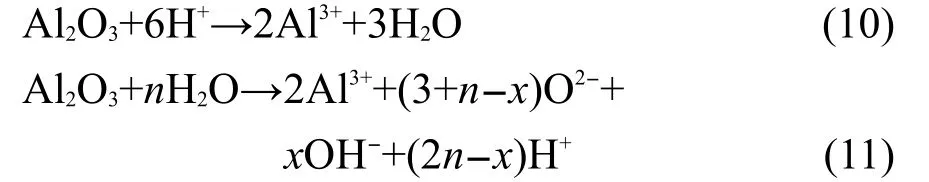
反应(11)式中n是参与水解反应中水的摩尔数与被溶解的Al2O3的摩尔数之比,x是水解反应中得到OH-的摩尔数.27,30奇怪的是,以上所有的电化学反应与电解液的组分(草酸、磷酸、己二酸铵、碳酸氢铵、次亚磷酸铵等等)没有直接关系,这些电解质组分主要是承担了电解液的导电任务.传统机理认为孔洞的形成是氧化铝被反应(10)酸性溶解的结果.但是,在阳极氧化过程中反应(10)中的H+离子要在电场作用下迁移到阴极释放氢气,因此电场导致的酸性溶解反应(10)在阳极发生的概率很小,这也是传统的孔道形成学说最大的局限性.30,32在非酸性的电解液中也能得到PAA孔道就是一个很好的例证,这说明(在不加外电场情况下)原溶液中是否存在H+离子(或浓度大小)与施加电场后孔道的形成没有直接关系.因此最近有研究者提出了孔洞形成和孔道加深的水解反应(11)的模式(原文中指出这种成孔模式对PAA和PATNT都适用),27不同研究者认为PAA形成机理的差异主要表现在过程(III)的不同,图7是PAA孔道传统发展模式与气泡模具效应成孔的对比示意图.
图7a是传统的PAA孔道加深模式.纳米孔洞形成以后在孔底部存在两个界面:电解液/氧化物(E/ O)界面和氧化物/金属(O/M)界面.氧化物的形成反应(7)发生的地点还存在争议,30-32一般认为反应(7)发生在O/M界面,而氧化铝被溶解和孔洞加深的反应(10)和(11)发生在E/O界面(图7a),依照“从上向下”的挖掘模式加深孔道,这个模式最大的缺陷是无法解释孔道底部的半球形和相邻元胞之间边界的形成过程,详细的分析参见综述文献.32而“气泡模具效应”的生长模型却主张孔壁的生长是围绕气泡模具“从下向上”生长的(图7b),Al2O3的形成反应(7)发生在E/O和O/M两个界面,当氧化铝生长到电子电流产生的临界厚度时,32反应(2)或(4)导致氧气析出,这时氧化物生长和氧气气泡增长同时进行,由于外界大气压(0.1 MPa)和电解液的压力使氧气气泡无法马上溢出,因此氧气气泡起到模具效应, Al2O3只好围绕“气泡模具”向上生长,即发生氧化铝的粘性流动(图7b,flow),这符合Skeldon等39,40采用示踪原子证明的氧化物从孔底部向两侧孔壁流动迁移的观点.当纳米孔道形成后,待O2气泡溢出后电解液渗入到纳米孔道内部,O2气泡继续在孔道底部析出,这时O2气泡的模具效应更加明显(因为O2气泡更难从纳米孔道中溢出),新形成的Al2O3由于体积膨胀在孔道底部围绕“气泡模具”向上生长,导致两侧的孔壁向上生长(图7b(II)),氧化铝孔壁围绕“气泡模具”向上生长的结果与传统理论“从上向下”的挖掘模式看似是相同的,实则有本质的区别.传统挖掘成孔的模式无法解释的半球形底部和相邻元胞之间边界用“气泡模具效应”能得到很好的解释,相邻元胞之间的边界是由于氧化铝抱紧各自孔道底部的“气泡模具”自然生长的结果,详细的解释参见文献32(与电场分布和自组织过程有关).当次亚磷酸根阴离子在电场作用下迁移到孔道底部与O2发生反应后(H2PO2-+O2=H2PO4-),导致气泡模具效应减弱,因而产生了图2和图4中的特殊的PAA孔道.假如按照图7a所示传统的孔道形成模式(反应(10)和(11)),图2、图4和图6中的孔道结构的变化是无法解释的.

图7 传统机理的孔道发展示意图(a);氧气气泡模具效应与PAA的迁移模型(b)Fig.7 Schematic diagrams of the traditional mechanism of the nanopore evolution(a);oxygen bubble mould effect and the flow model of porous anodic alumina(b) E/O:interface of electrolyte and oxide;O/M:interface of oxide and metal
两种截然不同的成孔模式用图7能示意性说明,但PAA机理研究的难点就在于无法用原位表征技术进行动态跟踪,44也不可能找到“气泡模具”的直接证据.为了进一步证明气泡模具效应的存在,本文创新性地采用密封阳极氧化的方式进行了实验.依据是有O2析出的反应会受到气压高低的影响,而传统成孔反应(10)或(11)都不应该受气压的影响.将抛光的铝条铆接在电容器盖板的正、负极上,将相应的电解液灌装到直径25 mm、高度40 mm的电容器铝壳中,用电容器组立机将铝壳和盖板封口后就得到了密封的阳极氧化装置.利用阳极氧化过程中阴极产生的大量氢气使铝壳中压力不断增大,阳极氧化的电流密度和时间不同,铝壳内的压力根据库仑定量可以定量估算.图8是在密封及常压(0.1 MPa)条件下阳极氧化后得到的阳极氧化膜形貌的SEM照片.图8(a,b)是在密封条件下,铝在2%磷酸溶液中恒电流阳极氧化不同时间得到样品的SEM照片.图8a和图8b对应的氧化时间分别是70 s(对应图1d中最高点)和130 s.从图8a中可见,在氧化膜表面并不是任何凹坑位置都能形成规则孔洞胚胎,我们认为1处和2处形貌的差异是“气泡模具”造成的,1处存在“气泡模具”才能形成规则的孔洞胚胎,孔洞底部的半球状十分明显;而没有“气泡模具”的2处就无法形成规则的孔洞,从图8a和图8c可见,表面不规则的凹坑数量远远大于断面规则的孔道数量.
随着氧化时间加长和内部压力的升高,气泡模具效应更加明显,图8b中三个规则孔道更加明显,“口小肚大”或“上细下粗”的规则孔道和半球形底部是气泡模具形成的,正如图7b(I)所示,这是由于在密封条件下氧气气泡受到的压力更大,气泡模具效应更明显的缘故(在常规气压下氧化时130 s短时间内很难找到这种半球形底部的结构).而图8a中的2位置就是没有气泡模具的凹坑,其形状就是“上粗下细”的腐蚀凹坑.因为在SEM照片中根本不可能找到气泡模具的直接证据,目前只能通过改变气压或者添加还原剂的方式来佐证,35,36,41而且从众多SEM照片中寻找旁证也是非常困难的事,必须结合相应的阳极氧化条件对SEM照片的细枝末节进行仔细的分析.例如图8c的SEM照片是在正常气压下(0.1 MPa)氧化后得到的,PAA的两个单元元胞所占据的位置大约在400 nm以下,其中1号孔道的最上端几乎还是密封的,最上层的表面也几乎看不到孔洞(图4(b,d)中也是这种情形),按照图7a传统的成孔模式是无法解释的.而在密封氧化的条件下,由于气泡在孔洞中停留的时间更长,因此图8b中的孔道直径更大一些,相邻的两个单元元胞占据的位置远远大于400 nm,这一观点从图8(d,e)中可以得到进一步验证.

图8 在密封(a,b,d,e)及常压(c)条件下氧化得到的阳极氧化铝膜的形貌Fig.8 Morphologies of the anodic alumina films anodized in a sealed case(a,b,d,e)and under normal pressure(c)(a,b,c)2%(w)H3PO4,(d,e)2%(w)H3PO4+6%(w)NH4H2PO2
图8(d,e)的SEM照片是铝在2%(w)H3PO4+ 6%(w)NH4H2PO2溶液中进行密封阳极氧化后得到的,其电解液与阳极氧化条件与图4(c,d)是一样的,区别仅在于图4(c,d)是在常压下进行的阳极氧化,而图8(d,e)是在全密封条件下进行的阳极氧化.从图8d可见最初形成的孔道1和孔道2最后被上层氧化膜完全覆盖,变成了彻底的致密膜.但图8d中每个单元元胞大小在300 nm左右,远远大于图4c中的情形.这种现象的发生用图7a的成孔模式无法解释,不管是反应(10)还是反应(11)的成孔模式,密封阳极氧化都不会使形成孔洞的反应停止,因此图8 (d,e)所示的致密膜是不可能形成的.唯一合理的解释是依据图7b(I)的成孔模式.在密封氧化的初始阶段,压力还较小,O2气泡析出,孔道形成,外界压力逐渐增大(阴极始终在释放氢气),还原剂更容易进入孔道把O2气泡吸收,气泡模具效应消失,最后氧化膜填充了原来的孔道.需要指出的是图8d中表面的裂缝是电镜制样时人为折弯造成的,与阳极氧化过程无关.图8d的SEM照片告诉我们更重要的信息是:Al2O3的形成反应(7)来源于Al3+向外迁移和O2-向内迁移,在E/O和O/M两个界面(图7a)都可能形成氧化铝,这个事实用图7a的传统成孔模式是无法解释的.按照传统理论Al2O3只能在O/M界面生长,在E/O界面是发生氧化铝的溶解反应(10)或者水解反应(11),因为传统理论认为Al2O3在O/M界面的生长和E/O界面的溶解必须达到相互平衡才保证了PAA孔底部阻挡层厚度不变.从图8(d,e)可见填充孔洞和覆盖孔洞的氧化铝更多是在电解液/氧化物(E/O)界面生成的,因为图8d中的1和2位置半球形底部没有消失,这就说明在氧化物/金属(O/M)界面生成的氧化铝很少,这是图7a的传统理论完全无法解释的,因为传统机理认为O2-向内迁移比Al3+向外迁移更容易.32
Patermarakis和Diakonikolaou31在2012年最新的机理研究文献中首次提出了Al3+向外迁移的速率远远大于O2-向内迁移的速率,颠覆了传统机理的观点.因此在氧化铝形成过程中Al3+离子迁移的数目远远大于O2-离子迁移的数目,31这样图8(d,e)中在电解液/氧化物(E/O)界面上生成更多的Al2O3就得到了合理的解释,同时也否定了图7a中只在O/M界面生成Al2O3的观点.此外,图8(d,e)的证据为今后研究阳极氧化过程中离子的迁移规律提供了一条崭新的思路.
此外,仔细分析图8e可以得到下列推论,图8e中的1、2、3和4号位置原先都有孔道,但1号孔道最长,其余三个孔道较短.孔道越长说明孔道底部的气泡模具效应越强,因为孔道越长,在没有被氧化物覆盖时,孔道底部的O2气泡越不容易溢出(图7b (II)).图8e中的1号孔道底部半球形最明显,其余三个孔道底部的半球形不明显,这一实验事实恰好佐证了上述观点.实际上,通常在制备PAA模板时最初孔道直径存在差异(气泡大小不同造成的)、孔道底部阻挡层厚薄不一致(如图8c)是十分常见的,但只要阳极氧化条件控制得当,最终都能得到自组织有序的PAA模板,关于PAA自组织过程的本质我们在文献中有详细的论述.32本文为了验证氧气气泡模具效应,采用了添加还原剂和密封阳极氧化等创新的方法,但没有对O2气泡在纳米孔道中的被吸收程度进行更精确和定量的控制,这一点需要结合自组织的本质进行更深入的研究,相信这些观点对精确调控PAA和PATNT的结构具有重要的指导意义.
4 结论
为了验证“氧气气泡模具效应”形成PAA有序孔道的观点,鉴于无法用原位表征的方法找到氧气气泡形成孔道的直接证据,本文从改变电解液的组分,添加强还原剂的方法从反面对氧气气泡模具效应进行了验证.无论在草酸还是在磷酸电解液中(都是形成规则有序PAA孔道的典型电解液),都证明了还原剂的加入对PAA孔道内部结构和表面形貌有重要影响,随着电解液中还原剂含量的增加,孔道的直径明显变小,无序性增大.这些明显的变化都是由于次亚磷酸根直接吸收O2所致(H2PO2-+O2= H2PO4-).而且在含有还原剂的电解液中制备的阳极氧化铝膜的导电性大大提高.在密封阳极氧化的条件下,次亚磷酸根能直接吸收掉孔道中的氧气,使气泡模具效应消失,得到完全的致密型氧化膜,结果表明在铝的阳极氧化过程中Al2O3更多是在电解液/氧化物界面上生成,这说明Al3+迁移的速率远远大于O2-迁移的速率.本文的实验事实从侧面证明了PAA中规则有序的纳米孔道是氧气气泡模具效应的结果.本文的研究思路为PAA和PATNT的结构调控提供了一种新的方法,同时为提高PATNT的导电性提供了有益的参考,这对提高PATNT的光电转换效率具有重要的实际应用价值.
致谢:感谢南京理工大学化工学院张常山和董伟教授在还原剂选择和电化学机理上的有益讨论,感谢南京春田电子有限公司无偿提供的铝箔和密封阳极氧化装置的制备,感谢宝鸡荣豪钛业有限公司无偿提供的高纯钛箔.
(1) Zhang,H.;Hu,Y.J.;Wu,P.;Zhang,H.;Cai,C.X.Acta Phys.-Chim.Sin.2012,28,1545. [张 华,胡耀娟,吴 萍,张 卉,蔡称心.物理化学学报,2012,28,1545.]doi:10.3866/ PKU.WHXB201203026
(2)Zhang,W.G.;Shang,Y.P.;Liu,L.N.;Yao,S.W.;Wang,H.Z. Acta Phys.-Chim.Sin.2011,27,900.[张卫国,尚云鹏,刘丽娜,姚素薇,王宏智.物理化学学报,2011,27,900.]doi: 10.3866/PKU.WHXB20110344
(3)Guo,Y.Y.;Wang,M.;Mao,X.B.;Jiang,Y.X.;Wang,C.;Yang, Y.L.Acta Phys.-Chim.Sin.2010,26,2037. [郭元元,汪 明,毛晓波,蒋月秀,王 琛,杨延莲.物理化学学报,2010,26, 2037.]doi:10.3866/PKU.WHXB20100734
(4) Santos,A.;Balderrama,V.S.;Alba,M.;Formentin,P.;Ferre-Borrull,J.;Pallarès,J.;Marsal,L.F.Adv.Mater.2012,24,1050. doi:10.1002/adma.v24.8
(5)Chen,W.;Jin,B.;Hu,Y.L.;Lu,Y.;Xia,X.H.Small 2012,7, 1001.
(6) Ding,J.N.;Zhu,Y.;Yuan,N.Y.;Ding,G.Q.Thin Solid Films 2012,520,4321.doi:10.1016/j.tsf.2012.02.030
(7)Dong,H.Q.;Pan,X.;Xie,Q.;Meng,Q.Q.;Gao,J.R.;Wang,J. G.Acta Phys.-Chim.Sin.2012,28,44.[董华青,潘 西,谢 琴,孟强强,高建荣,王建国.物理化学学报,2012,28, 44.]doi:10.3866/PKU.WHXB20122844
(8) Li,H.H.;Chen,R.F.;Ma,C.;Zhang,S.L.;An,Z.F.;Huang, W.Acta Phys.-Chim.Sin.2011,27,1017.[李欢欢,陈润锋,马 琮,张胜兰,安众福,黄 维.物理化学学报,2011,27, 1017.]doi:10.3866/PKU.WHXB20110514
(9)Wang,D.A.;Yu,B.;Wang,C.W.;Zhou,F.;Liu,W.M.Adv. Mater.2009,21,1964.doi:10.1002/adma.v21:19
(10) Li,D.D.;Zhao,L.;Jiang,C.H.;Lu,J.G.Nano Lett.2010,10, 2766.doi:10.1021/nl1004493
(11) Lin,J.;Chen,J.F.;Chen,X.F.Electrochem.Commun.2010, 12,1062.doi:10.1016/j.elecom.2010.05.027
(12) Lee,K.;Kim,D.;Roy,P.;Paramasivam,I.;Birajdar,B.I.; Spiecker,E.;Schmuki,P.J.Am.Chem.Soc.2010,132,1478. doi:10.1021/ja910045x
(13) Xu,X.J.;Fang,X.S.;Zhai,T.Y.;Zeng,H.B.;Liu,B.D.;Hu, X.Y.;Bando,Y.;Golberg,D.Small 2011,7,445.doi:10.1002/ smll.201001849
(14) Lee,W.;Ji,R.;Gösele,U.;Pippel,E.;Nielsch,K.Nat.Mater. 2006,5,741.doi:10.1038/nmat1717
(15) Lee,W.;Schwirn,K.;Steinhart,M.;Pippel,E.;Scholz,R.; Gösele,U.Nat.Nanotechnol.2008,3,234.doi:10.1038/ nnano.2008.54
(16)Ye,Q.M.;Song,Y.;Liu,P.;Hu,J.J.Prog.Chem.2011,23, 2617.[叶秋梅,宋 晔,刘 鹏,胡隽隽.化学进展,2011,23, 2617.]
(17) Li,S.Q.;Yin,J.B.;Zhang,G.M.Sci.China Chem.2010,53, 1068.[李仕琦,尹建波,张耿民.中国科学:化学,2010,53, 1068.]doi:10.1007/s11426-010-0155-3
(18) Lin,J.;Liu,K.;Chen,X.F.Small 2011,7,1784.doi:10.1002/ smll.v7.13
(19)Li,Y.;Ling,Z.Y.;Hu,X.;Liu,Y.S.;Chang,Y.J.Mater.Chem. 2011,21,9661.doi:10.1039/c1jm10781j
(20) Li,Y.;Ling,Z.Y.;Hu,X.;Liu,Y.S.;Chang,Y.Chem. Commun.2011,47,2173.doi:10.1039/c0cc04907g
(21) Bolger,C.T.;Fois,G.;Petkov,N.;Sassiat,N.;Burke,M.; Quinn,A.J.;Cross,G.L.W.;Holmes,J.D.Nanotechnology 2012,23,175602.doi:10.1088/0957-4484/23/17/175602
(22) Sulka,G.D.;Hnida,K.Nanotechnology 2012,23,075303. doi:10.1088/0957-4484/23/7/075303
(23)Cui,J.W.;Wu,Y.C.;Wang,Y.;Zheng,H.M.;Xu,G.Q.; Zhang,X.Y.Appl.Surf.Sci.2012,258,5305.doi:10.1016/ j.apsusc.2012.01.099
(24) Holubowitch,N.;Nagle,L.C.;Rohan,J.F.Solid State Ionics 2012,216,110.doi:10.1016/j.ssi.2012.03.016
(25) Romero,V.;Vega,V.;García,J.;Prida,V.M.;Hernando,B.; Benavente,J.J.Colloid Interface Sci.2012,376,40.doi: 10.1016/j.jcis.2012.02.066
(26) Roy,P.;Berger,S.;Schmuki,P.Angew.Chem.Int.Edit.2011, 50,2904.doi:10.1002/anie.201001374
(27)Su,Z.X.;Zhou,W.Z.J.Mater.Chem.2011,21,8955.doi: 10.1039/c0jm04587j
(28) Mazzarolo,A.;Curioni,M.;Vicenzo,A.;Skeldon,P.; Thompson,G.E.Electrochim.Acta 2012,75,288.doi:10.1016/ j.electacta.2012.04.114
(29) Regonini,D.;Satka,A.;Jaroenworaluck,A.;Allsopp,D.W.E.; Bowen,C.R.;Stevens,R.Electrochim.Acta 2012,74,244. doi:10.1016/j.electacta.2012.04.076
(30) Li,Y.;Ling,Z.Y.;Hu,X.;Liu,Y.S.;Chang,Y.RSC Adv.2012, 2,5164.doi:10.1039/c2ra01050j
(31) Patermarakis,G.;Diakonikolaou,J.J.Solid State Electrochem. 2012,doi:10.1007/s10008-012-1683-x
(32)Zhu,X.F.;Han,H.;Song,Y.;Duan,W.Q.Acta Phys.-Chim. Sin.2012,28,1291.[朱绪飞,韩 华,宋 晔,段文强.物理化学学报,2012,28,1291.]doi:10.3866/PKU. WHXB201204093
(33)Wang,F.;Wei,Q.S.;Zhang,Y.L.;Wu,K.;Xie,Y.C.Acta Phys.-Chim.Sin.2004,20,1134. [王 凡,卫庆硕,张玉玲,吴 凯,谢有畅.物理化学学报,2004,20,1134.]doi:10.3866/ PKU.WHXB20040915
(34)Yin,Y.X.;Jin,Z.G.;Tan,X.;Hou,F.;Zhao,L.Acta Phys.-Chim.Sin.2008,24,2133.[阴育新,靳正国,谭 欣,侯 峰,赵 林.物理化学学报,2008,24,2133.]doi:10.3866/ PKU.WHXB20081133
(35) Zhu,X.F.;Liu,L.;Song,Y.;Jia,H.;Yu,H.;Xiao,X.;Yang,X. L.Monatsh.Chem.2008,139,999.doi:10.1007/ s00706-008-0893-5
(36) Zhu,X.F.;Song,Y.;Liu,L.;Wang,C.;Zheng,J.;Jia,H.;Wang, X.Nanotechnology 2009,20,475303.doi:10.1088/0957-4484/ 20/47/475303
(37) Coz,F.L.;Arurault,L.;Datas,L.Mater.Charact.2010,61, 283.doi:10.1016/j.matchar.2009.12.008
(38)Gonzalez-Rovira,L.;Lopez-Haro,M.;Hungria,A.B.;Amrani, E.K.;Sanchez,J.M.;Calvino,J.J.;Botana,F.J.Corrosion Sci. 2010,52,3763.doi:10.1016/j.corsci.2010.07.027
(39) Skeldon,P.;Thompson,G.E.;Garcia-Vergara,S.J.;Iglesias-Rubianes,L.;Blanco-Pinzon,C.E.Electrochem.Solid St.2006, 9,B47.
(40) Garcia-Vergara,S.J.;Skeldon,P.;Thompson,G.E.;Habazaki, H.Electrochim.Acta 2006,52,681.doi:10.1016/j.electacta. 2006.05.054
(41) Zhu,X.F.;Song,Y.;Yu,H.D;Jia,H.B.;Yang,X.L.;Xiao,Y. H.;Lu,L.D.;Wang,X.Chinese Journal of Vacuum Science and Technology 2009,29,90. [朱绪飞,宋 晔,俞华栋,贾红兵,杨修丽,肖迎红,陆路德,汪 信.真空科学与技术学报,2009, 29,90.]
(42) Schwirn,K.;Lee,W.;Hillebrand,R.;Steinhart,M.;Nielsch,K.; Gösele,U.ACS Nano 2008,2,302.doi:10.1021/nn7001322
(43) Lee,W.;Scholz,R.;Gösele,U.Nano Lett.2008,8,2155. doi:10.1021/nl080280x
(44) Patermarakis,G.;Moussoutzanis,K.Electrochim.Acta 2009, 54,2434.doi:10.1016/j.electacta.2008.11.064
(45) Zhu,X.F.;Liu,L.;Zhao,B.C.The Chinese Journal of Nonferrous Metals 2003,13,1031. [朱绪飞,刘 霖,赵宝昌.中国有色金属学报,2003,13,1031.]doi:10.3321/j.issn: 1004-0609.2003.04.042
May 28,2012;Revised:August 10,2012;Published on Web:August 15,2012.
Influence of Reducer on the Nanopore Structure of Porous Anodic Alumina
ZHU Xu-Fei1,*HAN Hua2,*MA Hong-Tu2LU Chao1QI Wei-Xing1XU Chen1
(1Key Laboratory of Soft Chemistry and Functional Materials of Ministry of Education,Nanjing University of Science& Technology,Nanjing 210094,P.R.China;2National Engineering and Technology Research Center for ASIC Design, Institute of Automation,Chinese Academy of Sciences,Beijing 100190,P.R.China)
In recent years,attention has been focused on adjustment and control of the nanostructures of porous anodic alumina(PAA)and porous anodic TiO2nanotubes(PATNT).Because the formation mechanism of PAA and PATNT is still unclear,it is difficult to adjust the nanostructures of PAA and PATNT. To validate the novel viewpoint of the nanopore resulting from an oxygen bubble mold,an innovative chemical approach was used to adjust the PAA nanostructures.One successful approach is to use a reducer to absorb the oxygen bubbles in the nanopores.A novel anodic alumina film was obtained in a mixed solution of the reducer and oxalic acid.The influence of the reducer on the PAA nanostructures which formed in H3PO4solution was investigated in detail.The experimental results showed that the regularity and the diameters of the nanopores in the PAA decreased as the reducer content increased.The differences in the voltage-time curves between electrolytes with and without the reducer were analyzed quantitatively.The results showed that the conductivity of the anodic oxide film that formed in the electrolyte with the reducer was better than that in the electrolyte without the reducer.When aluminum anodizes in a sealed case,oxygen bubbles are easily absorbed by the reducer,the oxygen bubble mold effect disappears,and a compact alumina film is obtained.Overall,these results clearly demonstrate that nanopores result from the oxygen bubble mold effect.
Nanoporous material;Anodization;Electrolyte;Reducer;Formation mechanism
10.3866/PKU.WHXB201208151
∗Corresponding authors.ZHU Xu-Fei,Email:zhuxufei.njust@gmail.com;Tel:+86-25-84315949.
HAN Hua,Email:hua.han@ia.ac.cn;Tel:+86-10-62620080-666.
The project was supported by the National Natural Science Foundation of China(61171043,51077072)and National Science and Technology Major Project of the Ministry of Science and Technology of China(2009ZX01021-002).
国家自然科学基金(61171043,51077072)和国家科技重大专项资金(2009ZX01021-002)资助项目
O646
猜你喜欢
山东冶金(2022年4期)2022-09-14
内燃机与动力装置(2022年1期)2022-03-21
能源工程(2021年1期)2021-04-13
湖北农机化(2020年4期)2020-07-24
氯碱工业(2020年9期)2020-03-02
中学化学(2019年4期)2019-08-06
中学化学(2019年4期)2019-08-06
环境保护与循环经济(2017年4期)2018-01-22
筑路机械与施工机械化(2017年5期)2017-08-31
当代化工研究(2016年2期)2016-03-20