高压下β-HMX热分解机理的ReaxFF反应分子动力学模拟
2012-12-11 09:26:08周婷婷石一丁黄风雷
物理化学学报 2012年11期
周婷婷 石一丁 黄风雷
(北京理工大学,爆炸科学与技术国家重点实验室,北京100081)
高压下β-HMX热分解机理的ReaxFF反应分子动力学模拟
周婷婷 石一丁 黄风雷*
(北京理工大学,爆炸科学与技术国家重点实验室,北京100081)
采用ReaxFF反应分子动力学方法研究了不同压缩态β-HMX晶体(ρ=1.89,2.11,2.22,2.46,2.80, 3.20 g·cm-3)在T=2500 K时的热分解机理,分析了压力对初级和次级化学反应速率的影响、高压与低压下初始分解机理的区别以及造成反应机理发生变化的原因.发现HMX的初始分解机理与压力(或密度)相关.低压下(ρ<2.80 g·cm-3)以分子内反应为主,即N-NO2键的断裂、HONO的生成以及分子主环的断裂(C-N键的断裂).高压下(ρ≥2.80 g·cm-3)分子内反应被显著地抑制,而分子间反应得到促进,生成了较多的O2、HO等小分子和大分子团簇.初始分解机理随压力的变化导致不同密度下的反应速率和势能也有所不同.本文在原子水平对高压下HMX反应机理的深入研究对于认识含能材料在极端条件下的起爆、化学反应的发展以及爆轰等具有重要意义.
HMX;热分解; 压力;ReaxFF;分子动力学
1 引言
1,3,5,7-四硝基-1,3,5,7-四氮杂环辛烷(HMX)等硝胺类炸药因优越的爆轰性能而被广泛应用于武器装备及民用领域,这些炸药通过复杂的化学反应释放出巨大的能量.对化学反应过程的深入认识,如初始反应机理、详细的反应路径、反应产物的结构等,有利于建立宏观的燃烧和爆轰模型.Lewis等1应用密度泛函理论(DFT)对气相α-HMX分子分解过程的研究表明N-N键的断裂、HONO的离解、CN键的断裂及主环的分解是主要的反应路径.他们认为N-N键断裂生成NO2在单分子的初始分解中起着主要作用,而HONO的离解及C-N键的断裂在凝聚态HMX的分解过程中更有可能发生. Chakraborty等2通过DFT计算提出了单分子β-和δ-HMX分解的三种机理:N-N键断裂、HONO的离解、主环断裂生成CH2N2O2并进一步分解成CH2O和N2O.最近,Sharia等3应用DFT和过渡态理论(TST)研究了单分子和小体系HMX晶体(16个分子)的初始分解机理,得到了N-N键断裂、HONO离解及主环断裂的反应热和活化能.姜富灵等4采用DFT在B3LYP/6-31G(d)水平上研究了气相β-HMX分子N-NO2键断裂的焓变及自由能变.
对常压下凝聚态HMX热分解机理的研究有较多的实验报导.研究结果普遍认为在分解过程中生成了H2CO、N2O、HONO、HCN等中间产物,并进一步分解为N2、H2O、CO和CO2等最终产物.5-8Brill5提出了HMX分解的两种竞争机理,即HMX→4(HONO+HCN)和HMX→4(H2CO+N2O).Tang等6认为用多步反应来解释凝聚态HMX的热分解过程更为合适.Tarver等7提出了HMX热分解的三步模型:N-N键和C-N键的断裂生成初始产物;初始产物进一步分解为H2CO、N2O、HONO和HCN等中间产物;中间产物反应生成H2O、N2、CO和CO2等最终产物.
在压力作用下,凝聚态含能材料的分解过程更加复杂.因为压力会对含能材料的微观结构产生影响,进而影响化学反应.Gilman8认为,炸药在受到冲击作用时,强烈的压缩使炸药密度增大,共价键弯曲,当这种作用达到临界值时炸药发生局域金属化,导致电子激发.对共价分子,化学键的伸缩和弯曲使最高被占据分子轨道(HOMO)和最低空分子轨道(LUMO)之间的能隙降低.在分子晶体中,HOMO-LUMO带隙的闭合使得价带电子发生离域,导致化学反应的发生.9Margetis等10应用电荷自洽密度泛函紧束缚方法(SCC-DFTB)对静水和单轴压缩下凝聚态硝基甲烷(NM)能带结构的研究表明,C-N键的弯曲是造成带隙减小的主要原因,同时压缩作用使C-H键拉伸并最终造成氢质子的离解.Manaa11应用第一性原理研究了三氨基三硝基苯(TATB)带隙与硝基弯曲的关系,结果表明硝基弯曲会使带隙闭合,而硝基的弯曲可能是由剪切作用造成.Lu等12用DFT研究了静水压缩下β-HMX的结构和振动特性,发现压力显著地减小了N-N键长,说明N-N键在初始分解中起着重要作用. Kuklja等13,14应用DFT对剪切作用下FOX-7能带结构与态密度的研究发现,剪切作用使得带隙及化学反应的能垒降低,说明剪切作用可能是引发FOX-7热分解的主要原因.
在极端条件下(如高温高压),对凝聚态含能材料化学反应机理的研究因材料本身的性质、使用环境的特殊性以及反应的快速性而比较困难,特别是实验方面的研究.量子力学(QM)方法虽然能够准确地预测化学反应的过渡态、反应能垒、反应产物等,但主要用于研究单分子和小体系的静态性质.基于QM的分子动力学(MD)方法能够研究原子及分子结构的动力学过程,但因较大的计算量使得其在凝聚态含能材料特别是大体系上的应用受到限制. Manaa等15,16采用QM-MD方法研究了常压下单晶胞δ-HMX(6个分子)在温度为3500 K时的热分解过程,结果表明N-NO2键断裂是初始分解机理.最近,Zhu等17采用从头算MD方法研究了小体系β-HMX晶体(4个分子)在冲击作用下的化学过程,他们认为N-O键的断裂和主环的分解在初始分解中占主导地位.
基于第一性原理的ReaxFF反应力场18的发展为研究大体系的物理化学性质提供了有效的途径. ReaxFF反应分子动力学(ReaxFF-RMD)方法能够在较短的计算时间内模拟含有上百万个原子的体系在不同外加载荷下的物理化学过程,从原子和分子尺度提供详细和准确的信息.在含能材料领域, ReaxFF-RMD已经被广泛地用于研究热、压缩、冲击及剪切作用下的物理化学问题,为含能材料的化学反应机理提供了非常有价值的信息.采用这一方法,Rom等19研究了在一定温度范围内不同压缩态的液态NM的热分解机理,结果表明NM的初始分解机理与压缩量有关.Zhang等20研究了不同温度和密度下HMX和TATB晶体的热分解过程,得到了常压下初级和次级反应的活化能以及HMX与TATB热分解机理的主要区别.Strachan等21研究了不同密度环三亚甲基三硝胺(RDX)晶体的热分解过程,得到了各产物数量随密度的变化规律以及低密度与高密度晶体分解机理的主要区别.Strachan等22研究了RDX在不同冲击速度下的化学反应,结果表明冲击速度对RDX反应机理有较大影响.Zybin23、An24和Zhou25等分别研究了季戊四醇四硝酸酯(PETN)、RDX和HMX在压缩-剪切作用下材料的冲击各向异性,得到了与实验一致的研究结果.
在压缩或冲击作用下,炸药晶体被强烈压缩,分子间距显著减小,分子发生明显变形,使得化学反应过程比常压下的反应更为复杂,与气态单分子的分解更加不同.因此,研究高压下炸药的分解机理对于深入认识炸药在极端条件下的化学反应、冲击起爆以及爆轰等具有重要意义.尽管应用分子动力学方法对凝聚态HMX在压力作用下的化学反应已有报导,20但是却不够详细和深入,还有较多问题值得研究,如压力如何影响初始分解机理、反应路径及反应速率;常压和高压下反应机理的主要区别;反应机理发生变化的原因等.本文将应用ReaxFF-RMD方法研究不同压缩态β-HMX晶体的化学反应机理,以期解决以上问题.
2 模拟方法及说明
2.1 ReaxFF势函数
ReaxFF反应力场以键级(BO)为核心.18键级是原子间距离的函数,在分子动力学的每一次循环时进行计算.当化学键断裂时,与价键相关的能量和力变为零,因而该力场能够描述化学键断裂和生成.
ReaxFF势函数的表达式如式(1)所示.20它分成以下几个部分:基于键级的共价相互作用(Ebond、Eval和Etors)、原子间库仑力(ECoulomb)、分子间范德华作用力(EvdWaals)、氢键(EH-bond)以及各修正项(Elp、Eover、Eunder、Epen和Econj)用以正确描述不同化学环境下化学键的断裂和生成.
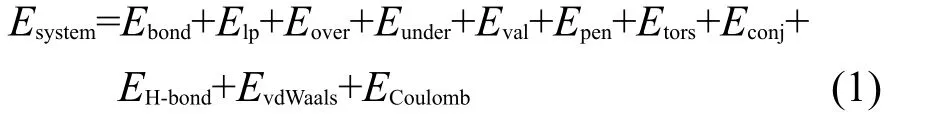
2.2 初始构象的构建
β-HMX晶胞来源于中子衍射晶体数据,26其分子及晶体结构如图1所示.首先构建4×2×3的超晶胞模型,共含有48个β-HMX分子、1344个原子.从低压到高压选取6个研究体系,晶胞参数来源于金刚石压腔实验27中压力p=0,2.5,4.6,10.6,26.0,42.6 GPa所对应的值,对应的密度分别为1.89、2.11、2.22、2.46、2.80和3.20 g·cm-3.
2.3 模拟过程说明
首先对模型进行几何优化,以获得合理的初始构型;对优化后的体系进行升温,并确保升温过程中晶体不分解;升温至目标温度T=2500 K后进行等温等容分子动力学模拟(NVT-MD).采用Berendsen方法对温度进行调节,使温度在设定值附近波动.本文所有的计算均采用周期性边界条件.模拟的时间步长为0.1 fs,模拟时间为20 ps.在20 ps内,主要分解产物已经全部出现且足以描述初级及次级化学反应以及压力对反应机理的影响.每隔50 fs记录一次原子坐标和速度以及每个原子对的键级,用以对分解产物进行分析.以BO=0.3作为产物生成与否的判据,当BO≥0.3时,认为化学键形成,产物生成.
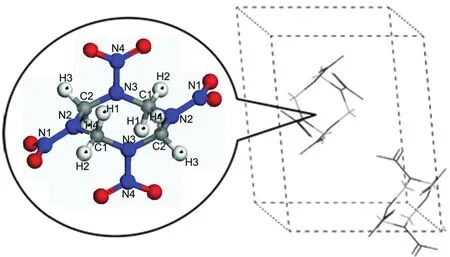
图1 β-HMX的分子结构及晶胞结构Fig.1 Molecular and crystal structures for β-HMX
2.4 反应速率分析方法
对不同压缩态HMX晶体在热分解过程中的反应速率分阶段进行分析.
(1)初始分解反应(吸热阶段):采用一阶衰减指数式(2)19对HMX分子数量随时间的变化进行拟合,得到不同压缩态HMX晶体热分解的初始反应速率.

式中,N0为HMX分子的初始数量;t0为HMX开始分解的时间;k1为初级反应速率.
(2)次级化学反应(放热阶段):对势能随时间的变化的分析发现,势能在反应的初始阶段存在一个最大值,对应的时间设为tmax.当t<tmax时,势能因体系吸热反应而升高;当t>tmax时,势能因体系放热反应而降低,因而tmax将反应分为吸热和放热两个阶段.采用一阶衰减指数式(3)19对放热阶段的势能随时间的变化进行拟合,得到不同压缩态HMX晶体热分解的次级反应速率.

式中,ΔUexo=U(tmax)-U∞,U∞为当t趋近于无穷大时势能的平衡值;U(tmax)为势能最大值;k2为次级反应速率.
(3)最终产物的生成.对不同压缩态HMX晶体热分解的最终产物随时间的变化采用式(4)进行拟合,得到最终产物的生成速率.在本文的模拟时间范围内(20 ps),最终产物还没有完全达到稳定值,因而本文没有对拟合得到产物的稳定值C∞进行讨论.
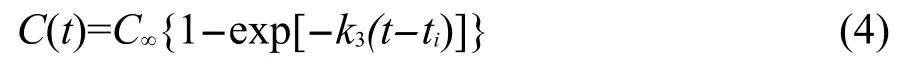
式中,C∞为t趋近于无穷时产物的稳定值;ti为产物开始形成的时间;k3为产物的生成速率.
3 模拟结果与讨论
3.1 反应速率
3.1.1 初始反应速率
不同压缩态晶体中HMX分子数量随时间的变化如图2所示,HMX分子数量随时间逐渐减少,在同一时刻,分子数量随着密度的增加先增加后减少,分界点为ρ=2.80 g·cm-3.采用式(2)对HMX分子数量随时间的变化进行指数拟合,得到初始分解速率k1随密度的变化,如图3所示.对ρ=1.89,2.11, 2.22,2.46,2.80,3.20 g·cm-3的体系,k1分别为8.13、6.80、5.13、2.04、7.52、26.32 ps-1,即当密度ρ<2.80 g· cm-3时,k1随密度的增加而减小,当密度ρ≥2.80 g· cm-3时,k1随密度的继续增加而增大.这说明HMX的初始分解速率与压力有关,低压会减慢HMX的初始分解速率,高压则会加快初始热分解.对ρ= 2.80,3.20 g·cm-3的高压缩态体系,对应的体积压缩量ΔV为32%和41%,即当ΔV≥32%时,HMX的初始分解速率反而随密度的增加而加快,这与Rom等19对不同压缩量的NM在T=2500-3500 K的温度范围内的热分解的研究结果相似.他们发现当ΔV<40%时,NM的初始分解速率降低;当ΔV≥40%时,初始分解速率反而加快.

图2 T=2500 K时不同压缩态β-HMX晶体中的HMX分子数量随时间的变化Fig.2 Evolution of HMX molecule number for β-HMX crystals with different densities at T=2500 K

图3 T=2500 K时不同压缩态β-HMX晶体的初始反应速率k1和次级反应速率k2Fig.3 Initial reaction rates k1and the second reaction rates k2for β-HMX crystals with different densities at T=2500 K
不同压缩态HMX晶体在分解过程中势能随时间的变化如图4所示,晶体的势能随密度的增加逐渐增大.随着密度的增加,晶体体积减小,原子间距和分子间距减小.原子间距的减少,分子内成键距离缩短,使得分子内原子间相互作用增强,势能增加.分子间距的减小,分子间的平衡距离被打破,分子间的排斥力占据主导地位,使得体系能量升高,势能增大.不同压缩态HMX的势能均随时间先升高后降低,最大值对应的时间为tmax.当t<tmax时,体系因初始分解反应而吸收能量,势能升高;当t>tmax时,体系因次级反应生成中间产物和最终产物而放出能量,势能降低.对ρ=1.89,2.11,2.22,2.46,2.80, 3.20 g·cm-3的体系,tmax分别为0.1、0.15、1.1、1.85、1.6和0.05 ps,说明低压下(ρ<2.80 g·cm-3)tmax随密度的增加而增大,吸热阶段延长;高压下(ρ≥2.80 g·cm-3) tmax随密度的继续增加而减小,吸热阶段缩短.tmax随密度的变化表明,当压力较小时,晶体密度较小(ρ<2.80 g·cm-3),体积较大,分子间还存在一定的可压缩空间,因此吸热过程可以持续一段时间.随着压力的增加,当晶体达到临界结构(ρ≥2.80 g·cm-3)时,分子间距很难再被压缩,此时体系热容过小,不再能够通过改变分子间距而吸收热量,而是直接发生分子间的放热反应,tmax变短.
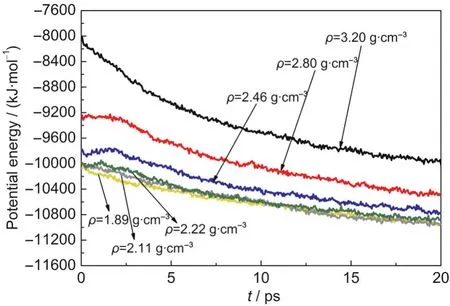
图4 T=2500 K时不同压缩态β-HMX晶体中平均每个HMX分子的势能随时间的变化Fig.4 Evolution of potential energy per HMX molecule for β-HMX crystals with different densities at T=2500 K
HMX的初始分解速率k1和势能达到最大值的时间tmax随密度的变化表明,HMX的初始分解机理在高压下(ρ≥2.80 g·cm-3)与低压下(ρ<2.80 g·cm-3)有所不同.常压下,分子内N-N键的断裂是最主要的初始分解机理;低压下,分子内N-N键的断裂仍然处于主导地位,但由于原子间距减小,键能增强, N-N键的断裂变得困难,导致初始反应变慢,吸热阶段延长;高压下,分子严重变形,分子间距及原子间距显著减小,使得分子间或分子内相邻非成键原子之间更容易接触而发生反应,导致较多小分子或大分子团簇的生成而迅速放出热量,吸热阶段缩短.Rom等19对不同压缩态NM的热分解的研究结果表明,在T≤3500 K时,低压下NM的初始分解机理以单分子分解为主,而高压下以分子间反应为主.他们将受激发分子的周围分子比作热浴,其作用是降低受激发分子的温度;对单分子分解而言,增加密度会增强激发分子与热浴之间的作用,使温度降低,从而降低受激发分子的分解速率;对双分子反应而言,增加密度会减少分子之间的空间,从而加快反应速率.因此,当温度不是非常高时,增加密度对不同的反应类型会有相反的影响,这与本文的研究结果相同.压力对HMX初始分解机理和反应路径的具体影响见3.2节.
3.1.2 次级反应速率
采用式(3)对不同压缩态HMX晶体的势能随时间的变化进行拟合,得到HMX分解过程中次级反应速率k2随密度的变化,如图3所示.对ρ=1.89, 2.11,2.22,2.46,2.80,3.20 g·cm-3的体系,k2分别为0.06、0.086、0.097、0.104、0.117和0.128 ps-1,即k2随密度的增加而增大,表明压力会加快次级反应速率.但k2并不是无限地增大,而是随着密度的增加趋于一个极值.Rom等19对不同压缩量NM热分解的研究也发现NM中间产物的反应速率随压缩率的增加而增加.压力减小了自由空间,使得初始分解产物更容易相互碰撞发生次级反应而生成中间产物和最终产物,次级反应速率加快.
3.2 产物分析
3.2.1 初始及中间反应产物
不同压缩态HMX晶体在T=2500 K时的主要分解产物(NO2、HONO、NO3、HNO3、HNO、NO、H2O、N2、CO2等)随时间的变化如图5所示.常压下,在HMX的热分解过程中观察到三种主要的初始分解机理:N-NO2键的断裂、HONO的离解和HMX分子主环的断裂(C-N键的断裂),这与前人的研究结果1-3一致.NO2是最主要的初始分解产物,即在HMX的初始反应中,N-NO2键断裂最容易发生. NO2的数量在达到最大值后迅速减小,这是由于次级反应消耗掉大量的NO2,生成NO3、HNO3等中间产物.HONO是仅次于NO2的初始分解产物,并进一步分解生成NO、HO、HNO等次级产物.HMX分子主环断裂的主要产物为C2H2O2N2,并迅速分解成HCN、HCON、H2CO、CON等中间产物.相比于NO2和HONO的数量,由主环断裂所生成的产物很少,说明主环断裂在HMX的热分解过程中不占主导地位,这与Sharia等3的研究结果相同.
Chakraborty等2采用B3LYP/6-31G(d)计算得到的气相HMX分子中N-NO2键断裂和HONO离解的能垒分别为166.52和186.61 kJ·mol-1.Sharia等3采用平面波密度泛函理论和广义梯度近似(GGA)得到的这两个反应的能垒分别为179.08和181.59 kJ·mol-1.他们考查了三种主环断裂模式,3即(1) C4H8N8O8→2C2H4N4O4,并进一步分解为两个CH2N2O2;(2)C4H8N8O8→CH2N2O2+open RDX;(3) C4H8N8O8→4CH2N2O2,对应的能垒分别为406.68、293.72和347.27 kJ·mol-1.3因此,N-N键断裂和HONO离解因较低的反应能垒比C-N主环断裂更容易发生.对凝聚态HMX晶体,Sharia等3认为主环断裂因较高的能垒及较慢的反应速率而不太可能出现,因而他们只计算了N-NO2键断裂和HONO离解的能垒,分别为200.41和218.82 kJ·mol-1.3所以N-NO2键断裂和HONO离解在HMX晶体的初始分解中占主导地位,且N-NO2键断裂最容易发生.

图5 T=2500 K时不同压缩态β-HMX晶体中平均每个HMX分子的主要分解产物随时间的变化Fig.5 Evolution of products per HMX molecule for β-HMX crystals with different densities at T=2500 K
压力对HMX晶体的初始分解机理会产生影响.从图5可以看出,初始分解产物NO2和HONO的数量随体系密度的增加而减少;高压下有较多的小分子生成,如O2、HO等,且数量与NO2和HONO相当.下面将详细分析密度对主要反应路径的影响.
3.2.1.1 N-NO2键断裂
压力对初始反应路径N-NO2键断裂的影响如图6(a)所示,NO2的数量随着体系密度的增加急剧减少.对ρ=1.89,2.11,2.22,2.46,2.80,3.20 g·cm-3的体系,NO2的最大值分别为1.6、1.25、1.2、0.8、0.5和0.3;在t=1 ps时,单个HMX分子中N-NO2键断裂的数量随密度的增加而减小,如图6(b)所示,说明压力显著地抑制了N-NO2键的断裂.Strachan等22对RDX在不同冲击速度下的分解机理的研究结果也表明当冲击速度较大时(vimp>8 km·s-1),NO2的数量反而随冲击速度的继续增加而降低.N-N键长随着体系密度的增大逐渐减小(如Supporting Information中表S1所示),键能增强,导致压力作用下NNO2键的断裂比较困难,NO2数量减少.Lu等12采用DFT对静水压缩下β-HMX的结构和振动特性的研究表明压力显著地减小了N-N键长.对反应路径的分析发现,压力作用下HMX分子中N-NO2键的断裂是一个可逆过程,即C4H8N8O8⇌C4H8N7O6+ NO2,N-NO2键断裂后形成的NO2很容易在较小的空间里再次与C4H8N7O6结合生成HMX,导致NO2数量的减少.且C4H8N7O6进一步分解产生NO2也更加困难,进一步造成NO2数量的减少.
NO3和HNO3的数量随着体系密度的增加而减少,如图6(c,d)所示.NO3主要从HMX分子脱落以及游离态NO2与O原子结合而成.在压力作用下, NO2数量急剧减少,导致它与O结合生成NO3的数量减小.HNO3主要为NO3与H原子结合以及NO2与HO结合而成,即NO3+H→HNO3和NO2+HO→HNO3.由于压力抑制了NO2和NO3的产生,导致在压力作用下HNO3的数量相应减少.
3.2.1.2 HONO离解
压力对HONO离解的影响与N-NO2键断裂类似,HONO的数量随密度的增加逐渐减少,如图7(a)所示.当ρ≥2.80 g·cm-3时,HONO的数量低于HNO3;对ρ=3.20 g·cm-3的体系,只有少量的HONO生成,其数量低于O2和HO.HONO主要由硝基(-NO2)上的O原子吸引-CH2上的H原子产生.2在压力作用下,HMX分子中二面角θ(H2-C1-N2-N1)和θ(H3-C2-N3-N4)随密度的增加逐渐增大(见Supporting Information中表S2所示),表明压力使-NO2和-CH2向两个相反的方向弯曲,增大了N-O…H-C之间的距离,从而降低了硝基与氢原子结合生成HONO的概率.特别是高压下(ρ≥2.80 g·cm-3)二面角增大明显,严重阻碍了硝基与氢原子的结合.另外,由于高压抑制了游离态NO2的产生,导致其与H原子结合生成HONO的概率减小,进一步造成HONO数量的减少.这与Strachan等22对RDX在不同冲击速度下的研究结果不同,他们发现HONO的数量随着冲击速度的增加而有所增加.

图6 T=2500 K时不同压缩态β-HMX晶体中平均每个HMX分子的分解产物NO2(a)、断裂的N-NO2键(b)、NO3(c)和HNO3(d)的数量随时间的变化Fig.6 Evolution of quantities of NO2(a),broken N-NO2bonds(b),NO3(c),and HNO3(d)per HMX molecule for β-HMX crystals with different densities at T=2500 K

图7 T=2500 K时不同压缩态β-HMX晶体中平均每个HMX分子的分解产物HONO(a)、NO(b)和HNO(c)的数量随时间的变化Fig.7 Evolution of quantities of HONO(a),NO(b),and HNO(c)per HMX molecule for β-HMX crystals with different densities at T=2500 K
高压对HONO离解的阻碍作用导致NO和HNO数量相应减少,如图7(b,c)所示.NO和HNO主要由HONO的分解产生:HONO→HO+NO,NO+ H→HNO.由于压力抑制了HONO的产生,造成NO减少,进而NO与H结合的概率减小,导致HNO的数量相应减少.对反应路径的分析发现,HONO的分解是一个可逆反应,即HONO分解产生的HO与NO会再次结合生成HONO:HO+NO→HONO,进一步导致NO和HNO减少.
3.2.1.3 分子主环断裂
HMX分子主环断裂的主要产物是H2CO、HCON和HCN.这些产物的数量较少,且随着体系密度的增加有所减少,特别是高压缩态体系,但是这些因主环断裂而生成的产物出现的时间却随着密度的增加有所提前,如Supporting Information中图S1(a-c)所示.这说明压力会加快HMX分子主环断裂的速率,但高压会抑制C-N键断裂后产物的进一步分解.在压力作用下自由空间减小,狭小的空间不利于环形大分子的存在,HMX分子受压变形,主环中二面角发生扭转,促进了C-N键断裂. Goto等28认为由压力引起的分子变形可以引发化学反应,因而在研究含能材料的点火起爆时不能忽视.HMX分子主环中二面角θ(N2-C2-N3-C1)和θ(N3-C1-N2-C2)随密度的变化最为明显(见Supporting Information中表S2),特别是对ρ≥2.80 g· cm-3的高压缩态体系,说明高压下分子主环变形严重使得N3-C1和N2-C2键容易断裂.HMX分子主环上的C-N键长随着密度的增大逐渐减小(见Supporting Information中表S1),但N3-C1和N2-C2键不同,对高压缩态体系键长反而增加,进一步说明N3-C1和N2-C2键在高压下容易断裂.这与Zhu等17对β-HMX晶体在冲击波作用下的化学过程的研究结果相同,他们认为主环的断裂是主要的初始分解路径之一.
3.2.1.4 大分子团簇的形成
与常压下相比,压缩态HMX体系在分解过程中容易形成大分子团簇,且大分子所含原子数随体系密度的增加而增加,这与Strachan等21对不同密度RDX晶体的热分解机理的研究结果相同.图8显示了在t=20 ps时生成的最大分子团簇中C、N原子数量随密度的变化,当ρ≥2.80 g·cm-3时,C、N原子数显著增加.常压下HMX分解程度高,分解出的分子质量较小;随着密度的增大,分子间距减小,自由空间减少,HMX分子相互挤压变形,C-N键断裂,显著的空间位阻效应使得C-N键断裂后易于发生分子间反应生成分子量很大的分子团簇,而不易于进一步分解成较小的分子,分解程度降低.
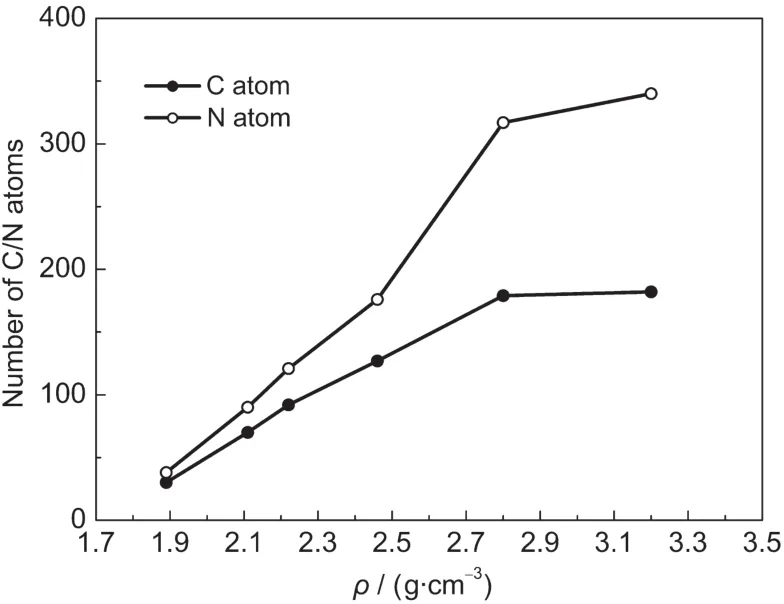
图8 T=2500 K时不同压缩态β-HMX晶体在t=20 ps时生成的最大分子团簇所含的C、N原子数Fig.8 Numbers of C and N atoms in the maximum molecule formed at t=20 ps for β-HMX crystals with different densities at T=2500 K
3.2.1.5 小分子的生成
压力促进了一些分子量很小的分子的形成,如O、H、HO、O2.随着体系密度的增加,O2和HO出现的时间提前,且数量增加,如Supporting Information中图S2所示.对ρ≥2.80 g·cm-3的高压缩态体系,O2和HO随时间的变化与NO2和HONO的对比见图9 (a-d).O2出现在HMX的初始分解阶段,与N-NO2键断裂的时间相同,早于HONO的生成;数量略少于NO2,与HONO相当.HO出现的时间略迟于NO2,与HONO相同;数量略少于NO2,与HONO相当甚至更多(ρ=3.20 g·cm-3).这说明在高压下(ρ≥2.80 g· cm-3),O2、HO等小分子的形成在HMX的初始分解机理中起着重要的作用,这与常压和低压下HMX的分解机理不同.在低压下(ρ<2.80 g·cm-3),HMX的初始分解以N-NO2键断裂和HONO离解等分子内反应为主,O2和HO等小分子很少出现.Strachan等22对不同冲击速度下RDX晶体的热分解机理的研究也表明由分子间反应生成的HO随冲击速度的增加而显著增加.
对化学反应路径的分析发现,高压下O、H主要是从HMX分子中直接脱离而成.在高压作用下, HMX分子受到挤压严重变形,HMX分子主环以外的部分C-H键和N-O键伸长,使得H、O原子比较容易脱落.不同压缩态HMX分子中的C-H键长和N-O键长随着密度的增加而减小,但是对高压缩态体系,C2-H4、N1-O1及N4-O3键反而伸长(如Supporting Information中表S1所示),说明这些键在高压下变得比较容易断裂,从而生成较多游离的H、O原子.Zhu等17采用从头算分子动力学方法对β-HMX晶体在冲击波作用下的化学过程的研究也表明,N-O键的断裂在初始分解中起着重要作用.Margetis等10应用SCC-DFTB方法对凝聚态NM在静水和单轴压缩下结构的研究发现,压缩作用使C-H键拉伸,最终造成氢质子的解离.

图9 T=2500 K时高压缩态β-HMX晶体中平均每个HMX分子的分解产物O2(a)、NO2(b)、HO(c)和HONO(d)的数量对比Fig.9 Comparisons of quantities of O2(a),NO2(b),HO(c),and HONO(d)per HMX molecule for β-HMX crystals with high densities at T=2500 K
这些游离小分子容易在较小的空间存在,且碰撞后进一步生成HO、O2等小分子.另外,高压下分子间距显著减小,分子主环外的相邻原子比较容易结合而生成HO、O2等小分子.对ρ=1.89,2.11,2.22, 2.46,2.80,3.20 g·cm-3的体系,分子间最近的H原子与 O原子间距分别为 0.2674、0.2586、0.2535、0.2423、0.2296和0.2073 nm,即分子间N-O…HC间距随着密度的增加而减小,有利于分子间H、O原子的结合.对反应路径的分析表明,HO主要来源于HONO的分解(HONO→HO+NO)以及H、O原子的直接结合(O+H→HO).高压抑制了HONO的产生,因此通过HONO分解产生的HO很少,大量的HO主要为后者产生.
3.2.2 最终产物的形成
HMX分解的最终产物主要是H2O、N2及CO2,如图5所示.随着体系密度的增加,H2O出现的时间逐渐推后,数量逐渐减少.对ρ=1.89,2.11,2.22,2.46, 2.80,3.20 g·cm-3的体系,H2O出现的时间分别为0.1、0.15、0.25、0.4、1.0和2.15 ps,最大值分别为2.0、1.85、1.9、1.6、1.25、0.75,如Supporting Information中图S3(a)所示.对不同压缩态体系生成的H2O随时间的变化按式(4)进行指数拟合,得到H2O的反应速率k3分别为0.061、0.056、0.050、0.045、0.032和0.016 ps-1,说明H2O的反应速率随着密度的增加逐渐减小,特别是高压下反应速率显著降低,说明压力不利于H2O的产生.对反应路径的分析发现H2O的产生与NO2的生成和HONO分解为HO和NO有关,即(1)C4H8N8O8→C4H8N7O6+NO2,(2)NO2+H→HONO,(3)HONO→HO+NO,(4)HO+H→H2O.NO2和HONO的数量随密度的增加逐渐减少,进而分解产生的HO和NO也相应减少,最终导致H2O的减少.且通过对反应路径的分析还发现,高压下HO+ H→H2O是一个可逆反应,即压力促进了H2O分解为HO和H,进一步造成H2O的减少.
N2的数量随密度的变化比较复杂,并不是随密度的增加而单调变化.当ρ<2.46 g·cm-3,N2的数量随着密度的增加逐渐增大;当ρ≥2.46 g·cm-3,N2的数量随密度的继续增大而减小,如Supporting Information中图S3(b)所示.对ρ=1.89,2.11,2.22,2.46,2.80, 3.20 g·cm-3的体系,N2的数量在t=20 ps时分别达到0.85、1.25、1.3、1.1、0.75和0.5,说明低压会促进N2的生成,而高压会抑制N2的产生.按式(4)拟合得到的不同压缩态体系生成N2的反应速率k3分别为0.027、0.040、0.042、0.035、0.021和0.018 ps-1,说明低压下反应速率随密度的增加而升高,高压下反应速率随密度的继续增加而降低.对反应路径的分析发现N2主要源于HMX分子主环的断裂生成C2N2O2及CHN2O2,进一步分解成CN2O和HN2O,最终生成N2.由于压力促进了主环的变形,加速了C-N键的断裂,而高压抑制了C-N键断裂后产物的进一步分解,最终导致低压下N2的数量随密度的增加而增加,而高压下随密度的增加而减少.N-NO2键断裂后产物的进一步反应也会生成N2.由于高压显著地抑制了N-N键的断裂,因而通过这一路径生成的N2也会减少.在不同冲击速度下,RDX热分解生成的N2随着冲击速度的增加而显著增加,22这与本文的研究结果不完全相同.
压力对CO2的影响与N2相似,但没有N2显著.当ρ<2.46 g·cm-3,CO2的数量随着密度的增加稍微有所增加;当ρ≥2.46 g·cm-3,CO2的数量随着密度的继续增加而有所减少,如Supporting Information中图S3(c)所示.对ρ=3.20 g·cm-3的体系,CO2的数量反而有所增加,这主要是由于高压下生成了较多的O和O2,使得C容易被氧化而生成较多的CO2.CO2是HMX主环断裂的最终产物,其随密度的变化说明压力对主环断裂的影响较为复杂,压力会加速CN键断裂的速率但高压会抑制C-N键断裂后产物的进一步分解,导致最终产物的减少.
4 结论
采用ReaxFF反应力场和分子动力学方法,研究了不同压缩态的HMX晶体在T=2500 K时的热分解过程.研究结果表明,HMX的初始分解机理与密度或压力有关,高压下(ρ≥2.80 g·cm-3)的初始热分解机理与低压(ρ<2.80 g·cm-3)下有所不同.常压下, HMX晶体热分解过程中三种主要的初始分解路径是N-NO2键的断裂、HONO的离解和C-N主环的断裂.低压下,HMX的初始分解机理以分子内NNO2键断裂和HONO离解为主,初始分解速率随密度增加而降低,体系以吸热反应开始,势能随时间先升高再降低.高压下HMX晶体的热分解程度降低,但分子间反应得到促进而生成了较多的HO、O2等小分子和大分子团簇,使得HMX单分子存在的时间变短,初始分解速率随密度增加而升高,体系主要以放热反应开始,势能随时间逐渐降低.高压缩态下HMX的初始热分解机理与低压缩态下最主要的区别是前者除部分分子内反应以外还有较多的分子间反应,而后者以分子内反应为主.次级反应速率随密度的增加有所增加,说明压力会加快HMX的次级反应.本文的研究工作有助于深入认识炸药在高温高压下的初始反应机理、反应速率、详细的反应路径,以及压力对化学响应的影响规律,这对于炸药的冲击起爆及极端条件下的化学反应研究具有借鉴意义和参考价值.
压力对HMX初始热分解机理的影响是否受温度的影响还有待研究.与RDX在不同冲击速度下的分解产物相比,静水压缩与冲击作用对含能材料的影响不完全相同;与HMX在一定冲击作用下的分解机理相比,高压缩态体系的分解机理与其有一些相似之处.因而有必要进一步研究凝聚态HMX在不同冲击作用下的化学反应,以得到静水压缩与冲击压缩对材料化学响应的影响及区别.
ReaxFF力场的发展为研究大体系的物理化学性质提供了非常有效的途径,且是目前主流的能用于研究凝聚态材料化学反应的力场.在含能材料领域,ReaxFF反应分子动力学方法得到了广泛而成功的应用,为凝聚态含能材料的化学反应提供了非常有价值的信息.当然也应该看到ReaxFF力场存在的不足.由于ReaxFF势函数比较复杂,力场参数较多,在通过拟合QM或实验数据得到力场参数时会产生一定的误差,且力场的参数化是一项具有较大难度和挑战性的工作,这使得ReaxFF的计算精度不如QM.除了力场参数,力场的函数形式也至关重要,比如对不在一个数量级的键能与分子间作用能的处理,不完善的函数形式可能会使计算结果不够准确.因此,进一步优化力场参数,发展函数形式是必要且有意义的工作.
Supporting Information Available: Evolution of quantities of H2CO,HCON,and HCN per HMX molecule,evolution of quantities of O2and HO per HMX molecule,and evolution of quantities of H2O,N2,and CO2per HMX molecule for β-HMX crystals with different densities at T=2500 K shown in Fig.S1, Fig.S2,and Fig.S3,respectively;bond length and dihedral an-gle in β-HMX molecule for crystals with different densities shown in Table S1 and Table S2,respectively,have been included.This information is available free of charge via the internet at http://www.whxb.pku.edu.cn.
(1) Lewis,J.P.;Glaesemann,K.R.;VanOpdorp,K.;Voth,G.A. J.Phys.Chem.A 2000,104,11384.doi:10.1021/jp002173g
(2) Chakraborty,D.;Muller,R.P.;Goddard,W.A.,III.J.Phys. Chem.A 2001,105,1302.doi:10.1021/jp0026181
(3) Sharia,O.;Kuklja,M.M.J.Phys.Chem.B 2011,115,12677.
(4) Jiang,F.L.;Zhai,G.H.;Ding,L.;Yue,K.F.;Liu,N.;Shi,Q. Z.;Wen,Z.Y.Acta Phys.-Chim.Sin.2010,26,409.[姜富灵,翟高红,丁 黎,岳可芬,刘 妮,史启祯,文振翼.物理化学学报,2010,26,409.]doi:10.3866/PKU.WHXB20100128
(5) Brill,T.B.J.Prop.Power 1995,11,740.doi:10.2514/3.23899
(6) Tang,C.J.;Lee,Y.J.;Litzinger,T.A.J.Prop.Power 1999,15, 296.doi:10.2514/2.5427
(7) Tarver,C.M.;Chidester,S.K.;Nichols,A.L.J.Phys.Chem. 1996,100,5794.doi:10.1021/jp953123s
(8) Gilman,J.J.Phil.Maga.B 1995,71(6),1057.doi:10.1080/ 01418639508241895
(9) Gilman,J.J.Phil.Maga.B 1993,67(2),207.doi:10.1080/ 13642819308207868
(10) Margetis,D.;Kaxiras,E.;Elstner,M.;Frauenheim,T.;Manaa, M.R.J.Chem.Phys.2002,117(2),788.doi:10.1063/ 1.1466830
(11) Manaa,M.R.Appl.Phys.Lett.2003,83(7),1352.doi:10.1063/ 1.1603351
(12) Lu,L.Y.;Wei,D.Q.;Chen,X.R.;Lian,D.;Ji,G.F.;Zhang,Q. M.;Gong,Z.Z.Mol.Phys.2008,106,2569.doi:10.1080/ 00268970802616343
(13) Kuklja,M.M.;Rashkeev,S.N.;Zerilli,F.J.Appl.Phys.Lett. 2006,89(7),71904.doi:10.1063/1.2335680
(14) Kuklja,M.M.;Rashkeev,S.N.Phys.Rev.B 2007,75(10), 104111.doi:10.1103/PhysRevB.75.104111
(15) Manaa,M.R.;Fried,L.E.;Melius,C.F.;Elstner,M.; Frauenheim,T.J.Phys.Chem.A 2002,106(39),9024.doi: 10.1021/jp025668+
(16)Manaa,M.R.;Fried,L.E.;Reed,E.J.J.Computer-Aided Materials Design 2003,10(2),75.doi:10.1023/B:JCAD. 0000036812.64349.15
(17)Zhu,W.H.;Huang,H.;Huang,H.J.;Xiao,H.M.J.Chem. Phys.2012,136,044516.doi:10.1063/1.3679384
(18) van Duin,A.C.T.;Dasgupta,S.;Lorant,F.J.Phys.Chem.A 2001,105(41),9396.
(19) Rom,N.;Zybin,S.V.;van Duin,A.C.T.;Goddard,W.A.,III; Zeiri,Y.;Katz,G.;Kosloff,R.J.Phys.Chem.A 2011,115, 10181.doi:10.1021/jp202059v
(20) Zhang,L.Z.;Sergey,V.Z.;van Duin,A.C.T.;Siddharth,D.; Goddard,W.A.,III.J.Phys.Chem.A 2009,113,10619.
(21) Strachan,A.;Kober,E.M.;van Duin,A.C.T.;Oxgaard,J.; Goddard,W.A.,III.J.Chem.Phys.2005,122(5),54502.doi: 10.1063/1.1831277
(22) Strachan,A.;van Duin,A.C.T.;Chakraborty,D.;Dasgupta,S.; Goddard,W.A.,III.Phys.Rev.Lett.2003,91(9),098301.doi: 10.1103/PhysRevLett.91.098301
(23) Zybin,S.V.;Goddard,W.A.,III;Xu,P.;van Duin,A.C.T.; Appl.Phys.Lett.2010,96,081918.doi:10.1063/1.3323103
(24)An,Q.;Liu,Y.;Zybin,S.V.;Kim,H.;Goddard,W.A.,III. J.Phys.Chem.C 2012,116(18),10198.doi:10.1021/ jp300711m
(25)Zhou,T.T.;Zybin,S.V.;Liu,Y.;Huang,F.L.;Goddard,W.A., III.J.Appl.Phys.2012,111(12),124904.doi:10.1063/ 1.4729114
(26) Choi,C.S.;Boutin,H.P.Acta Crystallogr.B 1970,26,1235.
(27) Yoo,C.S.;Cynn,H.J.Chem.Phys.1999,111(22),10229.doi: 10.1063/1.480341
(28)Goto,N.;Fujihisa,H.;Yamawaki,H.J.Phys.Chem.B 2006, 110,23655.doi:10.1021/jp0635359
June 4,2012;Revised:August 3,2012;Published on Web:August 3,2012.
Thermal Decomposition Mechanism of β-HMX under High Pressures via ReaxFF Reactive Molecular Dynamics Simulations
ZHOU Ting-Ting SHI Yi-Ding HUANG Feng-Lei*
(State Key Laboratory of Explosion Science and Technology,Beijing Institute of Technology,Beijing 100081,P.R.China)
The thermal decomposition mechanisms of condensed phase β-HMX at various densities(ρ= 1.89,2.11,2.22,2.46,2.80,3.20 g·cm-3)and at 2500 K were studied using ReaxFF reactive molecular dynamics simulations.The effects of pressure on the initial and secondary reaction rates,the main differences in the initial decomposition mechanisms between highly compressed and less compressed systems,as well as the reasons for these variations were analyzed.It was determined that the initial decomposition mechanisms of HMX were dependent on pressure(or density).At low densities(ρ<2.80 g· cm-3),intramolecular reactions are dominant,these being N-NO2bond dissociation,HONO elimination, and concerted ring fission by C-N bond scission.At high densities(ρ≥2.80 g·cm-3),intramolecular reactions are well restrained,whereas intermolecular reactions are promoted,leading to the formation of small molecules,such as O2and HO,and large molecular clusters.These changes in the initial decomposition mechanisms lead to different kinetic and energetic behaviors,as well as variations in the distribution of products.These results obtained through this work are significant in that they assist in understanding the chemical reactions involved in the initiation,reaction development,and detonation of energetic materials under extreme conditions.
HMX;Thermal decomposition;Pressure;ReaxFF;Molecular dynamics
10.3866/PKU.WHXB201208031
∗Corresponding author.Email:huangfl@bit.edu.cn;Tel:+86-10-68914518.
The project was supported by the National Natural Science Foundation of China(10832003).
国家自然科学基金(10832003)资助项目
O643;O642
猜你喜欢
特产研究(2024年1期)2024-03-12 05:40:56
人工晶体学报(2021年10期)2021-11-26 02:39:18
建材发展导向(2021年14期)2021-08-23 00:57:14
天然产物研究与开发(2019年10期)2019-11-05 10:12:44
中国煤层气(2019年2期)2019-08-27 00:59:30
环境与可持续发展(2017年2期)2017-04-06 03:07:30
火炸药学报(2014年1期)2014-03-20 13:17:27
机械制造与自动化(2014年1期)2014-03-01 04:21:57
少儿科学周刊·儿童版(2013年2期)2013-05-13 09:21:06
少儿科学周刊·儿童版(2013年2期)2013-05-13 09:21:06