
超高液相色谱法测定食品中甲醛含量
2012-12-02 00:58:22方桂红徐莉纪少凡
食品研究与开发 2012年9期
方桂红,徐莉,纪少凡
(1.海南医学院公共卫生学院,海南 海口 570101;2.海南省出入境检验检疫局技术中心,海南 海口 570311)
甲醛是一种重要的化工原料和有机溶剂,用途广泛。常用于有机合成、合成材料、涂料、橡胶、农药等行业,也可用作消毒、防腐和熏蒸剂。由于甲醛不仅对人体的嗅觉、呼吸道产生极大的刺激性和毒性作用,还具有潜在的致癌作用。美国环保局已证实甲醛是人类的一种致癌物质。因此,国家卫生部明确规定禁止使用甲醛或甲醛化合物作为食品添加剂使用。食品中甲醛的来源,一方面是食品生产原料自身含有的细胞代谢中间产物—“天然”甲醛,其含量因食品种类不同而有所差异。另一方面则是人为造成甲醛的污染:啤酒生产过程中添加甲醛以提高其稳定性,非法用于水产品以及其它干货产品的保鲜和防腐以及食品包装材料中甲醛残留对食品的污染,除此之外还有用含有甲醛的饲料喂养动物所造成甲醛在食品中的残留。这些污染已经严重损害了人类的健康,所以对食品中甲醛含量的检测是不容忽视的。
目前,食品中甲醛的分析方法已经很多,不同的食品种类处理方法也各不相同。常用的检测手段是分光光光度法、电化学和色谱法。分光光度法虽然操作简单,但容易产生假阳性。电化学方法操作繁琐,效率低。气相色谱法重现性和准确性不够好[1]。因此,本法基于甲醛溶于水的特点,采用水蒸气蒸馏后衍生化测定。该法无需对样品进行除蛋白等前处理,具有操作简便,灵敏度高,重现性好,衍生化后可直接进样等优点,在实际样品分析中可以得到较好的应用。
1 材料与方法
1.1 试剂与仪器
乙腈(GR):TEDIA 公司;磷酸(AR)、浓盐酸(AR):广州化学试剂厂;甲醛标准品(37%):SUPELCO公司。
UPLC液相色谱仪:美国Waters公司;A11型研磨机:德国IKA公司;2200 kjeltec自动蒸馏装置:美国FOSS公司;HWT-108水浴锅:天津恒奥技术发展有限公司;实验室用水均为哇哈哈纯净水。
1.2 溶液配制
2,4-二硝基苯肼:称取100 mg 2,4-二硝基苯肼溶解于24 mL盐酸中,加水定容至100 mL。
磷酸溶液:100mL磷酸中加入900mL水中,混匀。
1.3 色谱条件
色谱柱:AcquityUPLCBEHC18柱(100mm×2.1mm×1.7μm);柱温:40℃;流动相:乙腈-0.1%甲酸水(35+65);流速:0.3 mL/min;紫外检测器波长:365 nm;进样量:10μL。
1.4 样品前处理方法
1.4.1 样品的提取
将适量样品用研磨机捣碎,称取匀浆样品10.0 g(精确至0.01 g)于凯氏瓶中,准确加入20 mL蒸馏水,用玻璃棒搅拌均匀,浸泡30 min,加入10 mL磷酸溶液,水蒸气蒸馏约2 min,用已盛有20 mL水的比色管收集蒸馏液至100 mL,同时做空白对照实验。
1.4.2 衍生化反应
取蒸馏液5.0 mL,置于10 mL具塞比色管中,加入2mL2,4-二硝基苯肼和3mL乙腈,置60℃水浴20min,流水中快速冷却,直接吸取样液进行液相分析测定。
1.5 测定
1.5.1 甲醛标准储备溶液(3.33 mg/mL)的配制
吸取0.9 mL含量为37%甲醛标准溶液于100 mL容量瓶中,加水稀释至刻度,为甲醛标准储备液,冷藏保存。
1.5.2 甲醛标准工作液(100μg/mL)的配制
根据甲醛标准储备液的浓度,精密吸取适量于25 mL容量瓶中,加水至刻度,配置甲醛标准工作液,混匀备用,此液当日配置。
1.5.3 标准曲线的绘制
分别取 100μg/mL 的甲醛工作液 0.0、0.01、0.02、0.04、0.08、0.16、0.32 mL 于 20 mL 具塞比色管中,加蒸馏水至5.0 mL,按衍生化步骤操作,最后进行UPLC分析,然后以峰面积为纵坐标,甲醛的浓度为横坐标,绘制标准曲线。
2 结果与分析
2.1 前处理条件的选择
查阅相关文献发现,常见样品前处理手段主要采用直接加水振荡或超声提取、加入亚铁氰化钾等蛋白沉淀以及衍生化后反萃取[2-3]这3种方法。因此本文分别将上述3种方法和水蒸气蒸馏装置进行前处理比较。实验结果表明直接加水提取回收率较高但由于没有经过净化处理导致衍生化后样液污染检测系统,无法直接进样;在分光光度法中常用蛋白沉淀剂作为前处理方法,但测定结果稳定性差,精密度和灵敏度低,易产生假阳性;衍生化后用氯仿反萃取的方法虽提高灵敏度,可回收率不稳定,操作繁琐,耗时长,并且氯仿毒性大;然而本实验选用酸介质下水蒸气蒸馏,不仅可以避免直接蒸馏造成样品乳化现象的产生,可应用于各种基质,而且收集液澄清衍生化后直接进样分析,出峰效果好,无干扰峰,大大提高了检测的灵敏度。
考虑到蒸馏时间和酸介质对甲醛的回收率可能会造成影响,因此本文分别选取不同的蒸馏时间和磷酸溶液的添加量进行比较。实验结果显示蒸馏时间小于1 min时,检测限添加回收很难达到要求,可能是由于时间过短导致提取不完全,而蒸馏时间大于3 min时,回收率趋于稳定但蒸馏液过量降低了该方法的检测灵敏度,如图1所示,所以本文选取蒸馏时间约2min提取液100 mL。对于磷酸溶液添加量的确定,在查看相关文献基础上分别加入不同量进行蒸馏,结果表明加入量为10 mL时回收率能达到理想的效果,如图2所示。
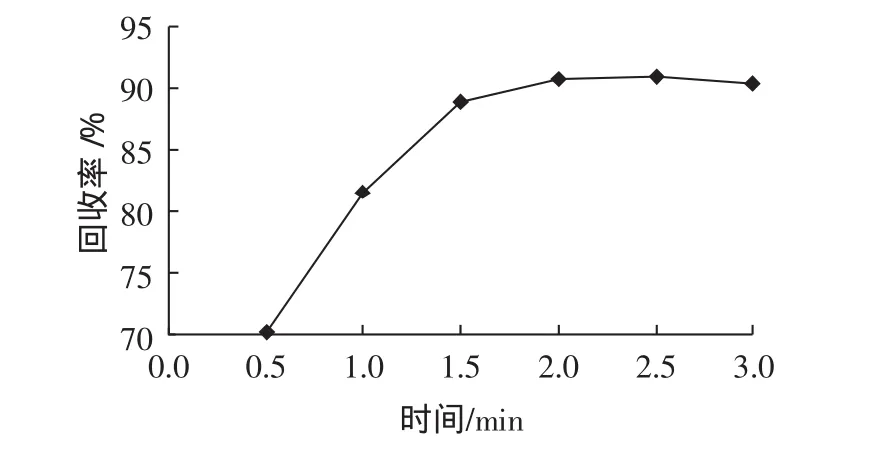
图1 不同蒸馏时间对回收率的影响Fig.1 The different distillation time to influence the recoveries
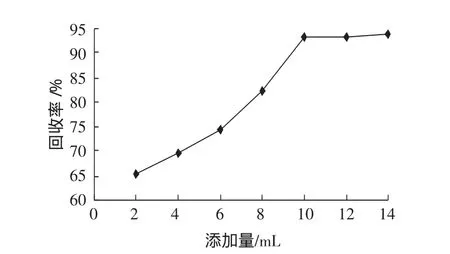
图2 不同磷酸溶液对回收率的影响Fig.2 The different phosphoric acid solution to influence the recoveries
2.2 测定条件的选择
实验过程中分别尝试使用Acquity UPLC BEH C18柱长为5 cm和10 cm两种色谱柱进行等度洗脱。分析测定结果发现短柱在使用时具有局限性,加大乙腈的比例,出峰时间过快导致与溶剂峰分不开,降低乙腈比例虽延长了出峰时间但较低浓度时易产生分裂峰,峰型不尖锐,无法定性定量,然而长柱出峰好且能通过调节流动相有效的排除干扰峰。因此10 cm柱的分离效果优于5 cm柱。在等度洗脱条件下,调节的乙腈-0.1%甲酸水溶液的比例发现两种溶液的配比为35∶65时效果最佳,加入甲酸的流动相使得峰型窄响应高,保留时间约为6 min,若加大乙腈的比例保留时间缩短但会导致峰拖尾。
根据文献资料,甲醛-2,4-二硝基苯腙的最大吸收波长分别在348、350、365 nm处,于是本文选择后两种波长进行分析检测并无明显差异,如图3、图4所示,因此最终确定紫外检测波长为365 nm。
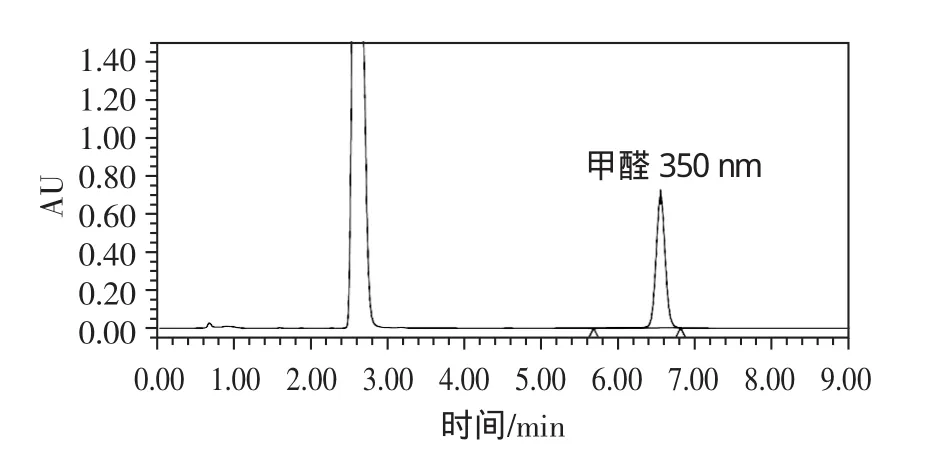
图3 波长为350 nm测定的甲醛标准谱图Fig.3 The formaldehyde chromatogram in wavelength 350 nm
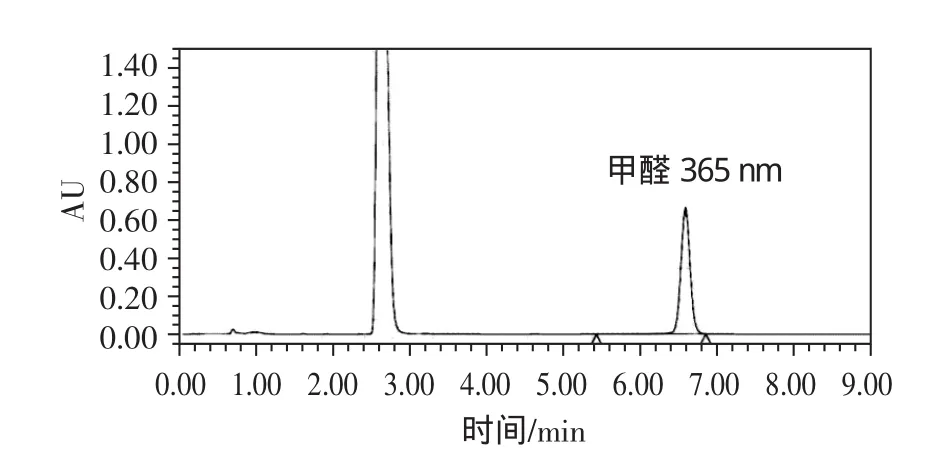
图4 波长为365 nm测定的甲醛标准谱图Fig.4 The formaldehyde chromatogram in wavelength 365 nm
2.3 线性关系及仪器检出限
根据1.5.3项下测定结果,得回归方程为Y=12068.1X+1337041;相关系数为0.9995。
结果表明,甲醛在0.050 mg/L~3.2 mg/L与峰面积呈现良好的线性关系,仪器的最小检出限(S/N=3)为0.050 mg/L。
2.4 不同样品加标回收和精密度实验
在选定的色谱条件下,对罗非鱼、鱼干、啤酒、腐竹和香菇五种样品进行添加回收实验,按照1.4样品前处理要求处理好样品待检。对样品连续分析6次,5种样品加标回收率和精密度实验结果见表1。
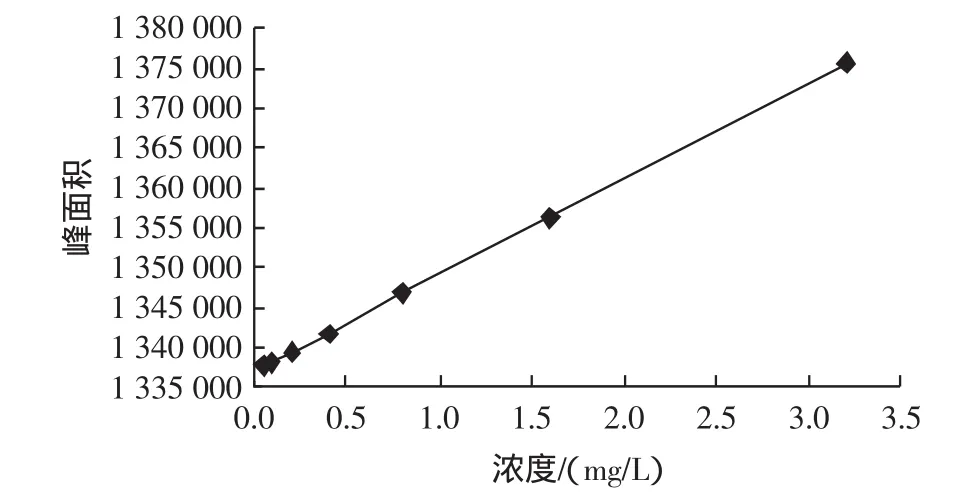
图5 线性关系考察Fig.5 Relationship between UPLC peak and formaldehyde concentration
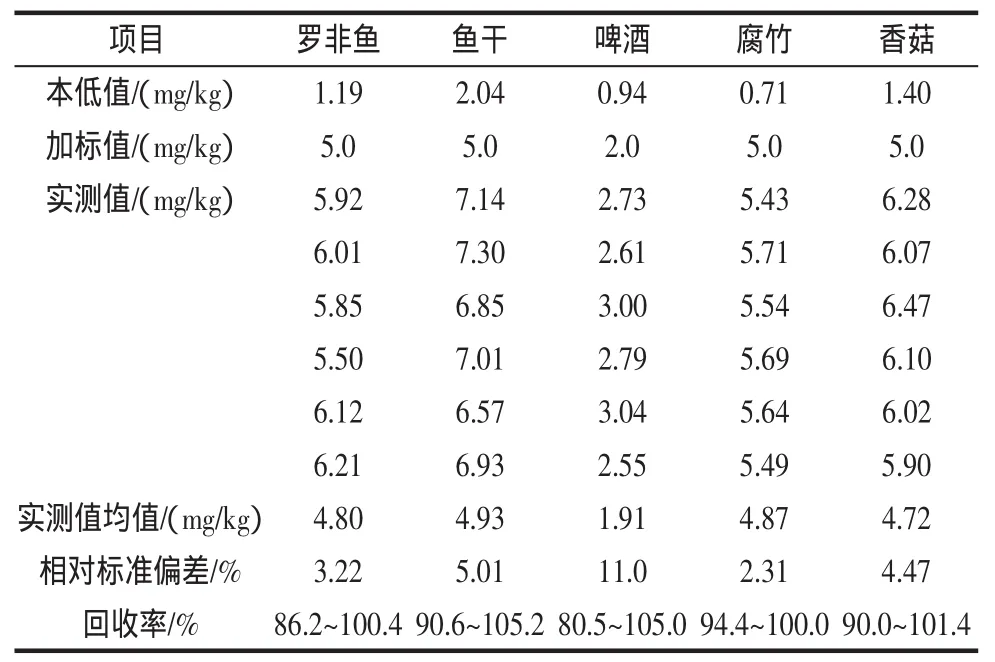
表1 样品加标回收和精密度实验(n=6)Table 1 The spiked recoveries and precision of sample
实验表明在选定的色谱条件下进样分析,甲醛的重现性好,精密度高,准确度好,样品回收率在80.5%~105.2%。
3 结论
本文选用磷酸溶液中水蒸气蒸馏法测定食品中甲醛的含量,相比于其它采用蛋白沉淀剂和直接提取的前处理方法应用范围广,适合于各种样品的分析,大大减少了基体对检测物质出峰的干扰,操作简便、快捷,避免使用有毒有害试剂。并且该方法还能有效的提高了甲醛的检测灵敏度和准确性,降低假阳性结果的出现,适用于食品中甲醛含量的测定。
[1]黎永艳,李必斌,张海霞,等.食品中甲醛的检验方法初探[J].广西预防医学,2006(1):54-55
[2]纳文娟,刘慧燕,方海田.食品中甲醛残留量检测方法的比较[J].保鲜与加工,2008,8(2):42-44
[3]彭科怀,陈江,赵年华,等.高效液相色谱法测定甲醛的研究[J].西南民族学院学报,2000,26(2):159-162
猜你喜欢
煤化工(2022年3期)2022-07-08 07:24:42
生物学通报(2021年4期)2021-03-16 05:41:26
江苏安全生产(2020年1期)2020-03-16 12:57:50
中国化肥信息(2019年12期)2020-01-16 08:40:06
中国化肥信息(2018年7期)2018-08-23 09:12:32
中国化肥信息(2018年6期)2018-08-23 09:11:42
中国化肥信息(2017年7期)2017-12-13 08:46:28
中国资源综合利用(2016年10期)2016-01-22 08:36:09
中央民族大学学报(自然科学版)(2014年3期)2014-06-09 08:54:31
中国质量与标准导报(2014年6期)2014-02-28 22:24:11
