
凝胶注模的凝胶反应动力学和均匀性
2012-11-29 10:33:48王小锋王日初彭超群罗玉林王志勇
中南大学学报(自然科学版) 2012年4期
王小锋,王日初,彭超群,罗玉林,王志勇
(1.中南大学 材料科学与工程学院,湖南 长沙,410083;2.中南大学 冶金科学与工程学院,湖南 长沙,410083)
在过去的几十年中,由于能够制备形状复杂且微观组织结构均匀的坯体,陶瓷胶态成型技术一直是研究的热点[1−4]。凝胶注模是一种重要的原位凝固胶态成型技术。除具有可成型复杂形状部件和坯体结构均匀等普通胶态成型的优点之外,它还具有坯体有机物含量低且强度高、工艺过程易控制和成本低廉等一系列优点[5]。该技术已用于 Al2O3[5,6],ZrO2[7],SiC[8]和 PZT[9]等各种氧化物或非氧化物陶瓷材料体系。目前,采用丙烯酰胺凝胶体系的水基凝胶注模因过程易控和适用性广等特点受到广泛关注[5−9],其原理为利用丙烯酰胺单体在悬浮液中进行凝胶反应形成具有三维网络结构的高分子物质,将陶瓷粉体颗粒原位固定,从而获得具有所需形状要求的制品。在凝胶注模的工艺过程中,悬浮液的原位固化即丙烯酰胺单体通过凝胶反应形成网络结构高分子的过程是十分重要的一道工序。Omatete等[6]研究悬浮液温度随凝胶反应时间的变化,并且指出凝胶反应的诱导期和凝胶点即凝胶反应开始发生的时间。马利国等[10]研究凝胶注模的固化过程,并分析陶瓷浆料凝胶点的影响因素。仝建峰等[11]研究表明可通过建立悬浮液温度与时间的曲线关系来研究悬浮液的凝固动力学。戴春雷等[12]甚至还研究凝胶注模的延迟固化。但上述研究均是以悬浮液中的凝胶反应为研究对象进行的。实际上,因为悬浮液的变化如陶瓷粉体材料体系的改变、粘度的变化和起皮抑制剂如聚乙二醇[13]的加入等对凝胶反应、坯体强度和均匀性等都会产生影响,所以,水溶液中的凝胶反应和凝胶均匀性才是凝胶注模的基础,很有必要对其进行研究。本文作者研究各工艺条件如引发剂浓度、催化剂浓度、单体浓度、单体与交联剂的质量比和起始反应温度等对凝胶反应和凝胶均匀性的影响。此外,还研究悬浮液固相体积分数对凝胶反应的影响。
1 实验材料与方法
1.1 原料
选用湖南水口山有色金属集团有限公司生产的氧化铍粉体为原料,平均粒径为0.5 μm。有机单体和交联剂分别为丙烯酰胺(AM)和 N,N’-亚甲基双丙烯酰胺(MBAM)(上海国药集团有限公司);引发剂为过硫酸铵(APS,长沙分路口化工厂);催化剂为N,N,N',N'-四甲基乙二铵(TEMED,北京化学试剂公司);分散剂为聚丙烯酸铵(日本东亚合成有限公司);去离子水为实验室自制水。
1.2 实验过程
精确称取APS配制质量分数为10%的溶液待用。称取一定量的单体AM和交联剂MBAM加入去离子水中,经搅拌均匀直至溶液透明,得到单体水溶液。在单体水溶液中加入分散剂NH4PAA和BeO粉体,球磨24 h制得固相体积分数为45%的BeO粉体悬浮液。
在单体水溶液中加入一定量的催化剂TEMED和引发剂APS溶液,静置;该溶液经一定时间反应后转变为凝胶。为了排除因氧气存在而产生“氧阻聚”的影响,将溶液在反应前抽真空处理,除去其中的氧气。
将BeO粉体悬浮液真空除气后,在设定温度下加入引发剂和催化剂并注入模具中,使悬浮液凝胶固化。
1.3 测试与表征
丙烯酰胺单体的自由基聚合过程是一个放热反应,其聚合热为82.8 kJ/mol[14],放热引起的温度变化比较明显。因此,采用聚合反应的热效应,即温度变化来表征单体聚合的转化率和聚合速度[12]。将一定量的单体水溶液或悬浮液加入烧杯内,然后将之置于可控温的保温容器内,待温度达到设定的温度后,加入引发剂APS或催化剂TEMED引发反应。同时,将温度探头插入反应溶液中,监测并记录反应体系温度随时间的变化。
将合成后的凝胶浸泡至溶胀平衡,切成长×宽×高为20 mm×10 mm×5 mm的小块,置于40 mm×10 mm×5 mm的比色皿中,再注入蒸馏水。用721B型可见分光光度计,在波长为580 nm时测定凝胶的透射比[15]。
2 实验结果
2.1 引发剂体积分数对凝胶反应和凝胶透射比的影响

图1 引发剂体积分数对凝胶反应的影响Fig.1 Effect of initiator concentration on gelation
图1所示为不同引发剂体积分数时凝胶反应的温度变化和对应的诱导期与反应期。由图1可知:随着引发剂体积分数的增加,凝胶反应诱导期持续缩短。此外,凝胶反应“S”曲线的斜率也逐渐增加,表明聚合速率也升高,因此,反应期也缩短。
图2所示为引发剂体积分数对合成凝胶透射比的影响。由图2可知:随引发剂体积分数的增加,凝胶透射比先增加而后减小,但总的来说引发剂体积分数对合成凝胶透射比的影响不大。
2.2 催化剂体积分数对凝胶反应和透射比的影响
图3所示为不同催化剂体积分数时凝胶反应的温度变化和相应的诱导期与反应期。由图3可知:随着催化剂体积分数的增加,诱导期和反应期均减小。
图4所示为催化剂体积分数对凝胶透射比的影响。由图4可知:随着催化剂体积分数的增加,凝胶透射比整体上逐渐减小,但总的来说对合成凝胶透射比的影响也不大。
2.3 单体质量分数对凝胶反应和透射比的影响
图5所示为单体质量分数对凝胶反应的影响和对应的诱导期与反应期。由图5可知:随着单体质量分数的增加,曲线的斜率也逐渐增加,表明增加单体质量分数能够促进聚合反应,使聚合反应速率增加。此外,各反应的最高温度也基本随单体质量分数的增加而增加。单体质量分数的增加使得参与反应的乙烯基浓度增加,而该反应为一个放热反应,因此,反应的最高温度也必然增加。反应期和诱导期随单体质量分数的增加而缩短(如图5(b)所示),即单体质量分数的增加提高了引发速率。
图6所示为单体质量分数对凝胶透射比的影响。由图6可见:单体质量分数过高或过低,凝胶的透明性均会降低,表明凝胶的结构均匀性变差。

图2 引发剂体积分数对合成凝胶透射比的影响Fig.2 Effect of initiator concentration on transmittance of gel

图3 催化剂体积分数对凝胶反应的影响Fig.3 Effect of catalyst concentration on gelation

图4 催化剂体积分数对凝胶透射比的影响Fig.4 Effect of catalyst concentration on transmittance of gel
2.4 单体与交联剂质量比对凝胶反应和透射比的影响

图5 单体质量分数对凝胶反应的影响(引发剂和催化剂体积分数分别为6 μL/100 mL和 3 μL/100 mL)Fig.5 Effect of monomer concentration on gelation
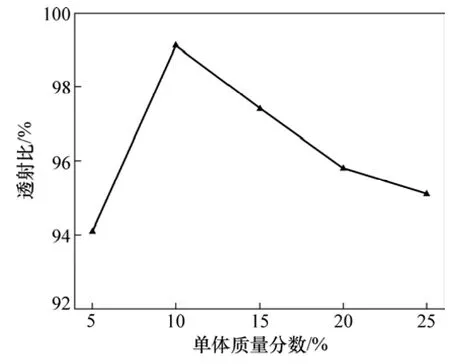
图6 单体质量分数对合成凝胶的透射比的影响Fig.6 Effect of monomer concentration on transmittance of gel
图7所示为单体比例对凝胶反应的影响和对应的诱导期与反应期。由图7可知:随着单体与交联剂质量比(m(AM)/m(MBAM))比例增加,凝胶反应的诱导期和反应期均缩短。图7还表明:凝胶聚合反应速率也体现在反应热效应(最高温度)上。当m(AM)/m(MBAM)为10∶1时,聚合反应放热产生的热积累使溶液的温度升高23.6 ℃;而当m(AM)/m(MBAM)为35∶1时,溶液的温度升高20.2 ℃,两者温度相差3.4 ℃。此外,图7中各“S”曲线中间段的斜率均十分相近,也表明在不同m(AM)/m(MBAM)的条件下,凝胶聚合反应速率相差不大。另外,与单体质量分的影响(图5)比较,m(AM)/m(MBAM)对凝胶反应的影响比单体浓度的影响小许多。显然,m(AM)/m(MBAM)对反应诱导期和反应期的影响也不大(如图7(b)所示)。

图7 m(AM)/m(MBAM)对凝胶反应的影响Fig.7 Effect of m(AM)/m(MBAM)ratio on gelation
图8所示为不同m(AM)/m(MBAM)下合成凝胶的透射比。由图8可以看出:随着m(AM)/m(MBAM)的降低即交联剂浓度的增加,凝胶透明性有所降低。由图8还可知:交联剂浓度对凝胶透射比的影响远大于单体AM浓度的影响因素,而且在单体比例为10∶1与15∶1之间(交联剂浓度分别为0.097和0.065 mol/L)存在突变。

图8 m(AM)/m(MBAM)对合成凝胶的透射比的影响Fig.8 Effect of m(AM)/m(MBAM)ratio on transmittance of gel
2.5 起始反应温度对凝胶反应和透射比的影响
图9所示为起始反应温度对凝胶反应的影响和对应的诱导期与反应期。从图9可见:随起始反应温度的增加,凝胶反应的诱导期和反应期均缩短;随起始反应温度的增加,反应从开始到结束的时间即持续的时间逐渐减少。在起始反应温度为40 ℃的条件下,反应持续的时间仅为30 s左右。
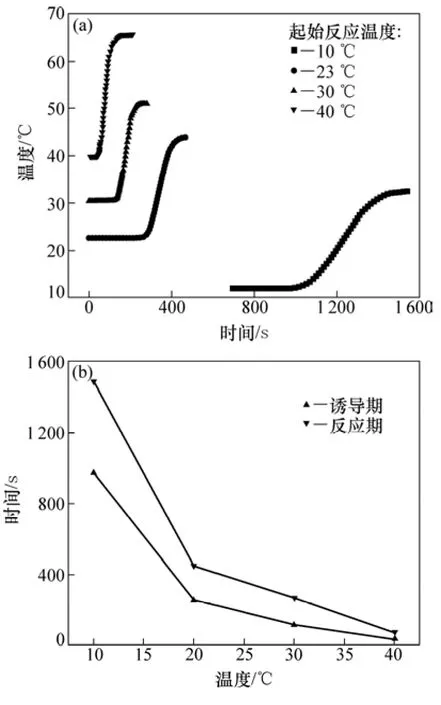
图9 起始反应温度对凝胶反应的影响(引发剂浓度和催化剂体积分数分别为6 μL/100 mL和2 μL/100 mL)Fig.9 Effect of initial reaction temperature on gelation
图10 所示为在不同起始反应温度条件下制备凝胶的透射比。由图10可知:随起始反应温度的增加,透射比先增加而后又小幅度下降。
2.6 固相体积分数对凝胶反应的影响
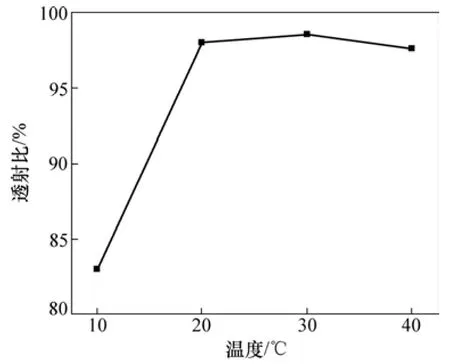
图10 起始反应温度对凝胶透射比的影响Fig.10 Effect of initial reaction temperature on transmittance of gel

图11 固相体积分数对凝胶反应的影响Fig.11 Effect of solids loading on gelation
图11所示为不同固相体积分数的悬浮液中凝胶反应体系温度随时间的变化曲线。从图11可见:悬浮液固相体积分数对凝胶反应的影响很大;随固相体积分数的增加,凝胶反应诱导期缩短,而反应期却小幅度延长(如图11(b)所示),表明凝胶反应的引发速率升高。
3 讨论
3.1 凝胶反应的基本原理
凝胶注模的工艺过程为:首先通过球磨和超声振荡等方式配成含有机单体AM和交联剂MBAM的高固相体积分数的粉体悬浮浆料,然后加入引发剂与催化剂等可使有机单体发生凝胶反应的物质,充分搅拌均匀后,将悬浮液注入模具中,最后在一定的温度条件下经过一段时间后凝胶反应被引发,AM与MBAM相互反应形成具有一定强度和柔韧性的三维网状结构,从而获得具有一定形状的坯体。凝胶注模的核心原理是采用形成的具有三维网络结构的高分子物质将分散均匀的粉体悬浮液中的颗粒包裹使之原位固定。显然,在整个工艺过程中,三维网络结构高分子的形成,即悬浮液中AM与MBAM的凝胶反应,具有非常关键的作用。根据高分子原理,AM与MBAM之间的反应为典型的自由基聚合反应。一般地,自由基聚合反应分为链引发、链增长和链终止3步基元反应[14,16]。链引发:链引发是形成单体自由基的反应。本研究采用引发剂过硫酸铵(APS)和催化剂(TEMED)引发反应。链引发分为2步反应:首先水溶液中的APS在热作用下均裂分解形成具有很高活性的初级自由基;然后它分别将AM和MBAM分子中的烯键打开并与之加成,形成单体自由基。链增长:所得单体自由基打开烯类分子的π键,加成,形成新自由基。新自由基的活性并不衰减,继续与烯类单体连锁加成,形成结构单元更多的链自由基。链终止:随着反应的进行,水溶液中的自由基浓度迅速增加。高活性的自由基彼此间易相互作用而终止,形成稳定的聚合物分子。
3.2 凝胶反应动力学
凝胶反应过程中,因为单体聚合时会放出能量,从而引起反应体系温度的升高,所以整个过程中温度随时间的增加而升高。忽略环境与反应体系间的传热关系,可采用反应体系温度随时间的变化关系来表征凝胶反应速率的变化[12,16]。在自由基聚合的反应中,链引发是最慢的一步,控制着总的聚合速率。引发剂浓度是影响速率和分子量的关键因素,对凝胶反应有很大影响。引发剂分解后,一部分用来引发单体聚合,另一部分则因诱导分解或笼蔽效应而消耗。诱导分解是一种自由基向引发剂的转移反应。笼蔽效应则是处在单体或溶剂“笼子”中的引发剂分子无法及时扩散出来,在笼内发生副反应形成稳定分子而消耗的现象。当引发剂浓度较低时,由于受到诱导分解和笼蔽效应的影响,引发剂分解产生的自由基很容易被损耗,没有足够多的引发剂自由基和单体作用形成单体自由基,进而进行链增长反应。随引发剂浓度的增加,引发剂自由基增多,单体自由基也随之增加,凝胶反应能够被完全引发,因此诱导期也随着引发剂浓度的增加而缩短,如图5(b)所示。并且增加引发剂浓度的效果十分明显,因此图5(b)中曲线较陡峭。当引发剂达到一定浓度后,产生的自由基足够且远远大于链增长反应的所需。因此,多余的自由基并没有参加链增长反应,相应地就不能更有效地缩短诱导期和反应期,表现在图5(b)中为曲线趋于平缓。
催化剂在反应过程中起到加速自由基产生和降低反应活化能的作用,因此,催化剂的加入必然使诱导期和反应期缩短(图3)。另外,催化剂浓度较小时,其作用十分明显;而催化剂的加入量已经较多时,其起到的加速作用则不再明显。凝胶反应中对诱导期和反应期起主要作用是溶液内自由基的量。少量的催化剂与引发剂反应能降低其生产自由基的激活能,因此,作用明显;当催化剂过量时,由于引发剂已几乎被全部消耗,自由基产生的量不再增加,过量的催化剂所起的作用没那么明显。
在某些情况下,除引发剂浓度外,单体浓度对链引发速率也有影响。单体浓度越高,初级自由基与单体结合进行链引发的概率也就越高,凝胶反应发生得越快。单体浓度与引发速率存在如下关系[16]:

相应地,与聚合速率存在如下关系:

式中:Ri,Rp,I,M,kd,kt和f分别代表反应速率、增长速率、引发剂浓度、单体浓度、引发速率常数、终止速率常数和引发剂效率。
式(1)和式(2)表明:增加单体浓度,引发速率和聚合速率均相应地升高。因此,凝胶反应的诱导期和反应期均缩短(图5)。
单体与交联剂质量比对凝胶反应也有影响。与单体浓度的影响原因相同,m(AM)/m(MBAM)增加,即MBAM浓度下降,使得溶液中的乙烯基浓度降低,因此,聚合反应速率稍微降低,凝胶反应的诱导期和反应期均缩短。
起始反应温度对凝胶反应起着十分重要的作用。Gelfi等[17]的研究表明:只有当起始反应温度高于某一温度之后,凝胶反应才能进行。起始反应温度的增加使得引发剂分解速率增加,自由基浓度升高,引发速率增加,因此,凝胶反应的诱导期缩短(图9)。
引发剂分解产生自由基是一个热激活的过程,满足阿伦尼乌斯方程:

其中:r为引发速率,R为气体参数,Ea为引发剂分解产生自由基的激活能。因此,采用图9中的数据,根据方程(3)可计算引发剂分解激活能,如图12 所示。从图12可见:反应激活能为79.45 kJ/mol,该值与其他文献报道的结果基本一致[6]。
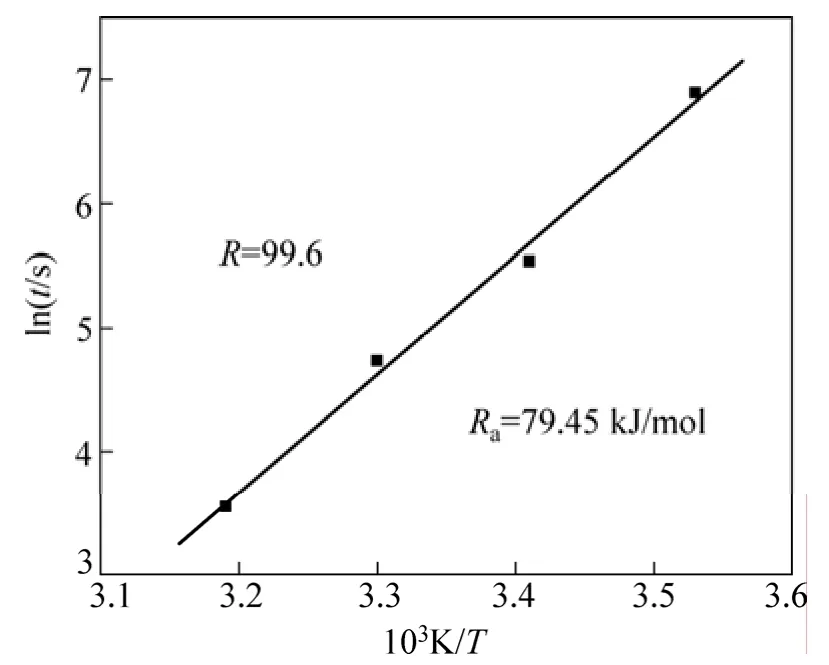
图12 引发剂分解激活能的计算Fig.12 Calculation of active energy of decomposition of initiator
增加悬浮液的固相体积分数能促进凝胶反应的进行。凝胶反应是靠溶液中的自由基引发而进行,并且自由基浓度直接决定凝胶反应速率。引发剂形成的自由基,因悬浮液黏度较高而无法快速扩散,从而造成局部引发剂浓度过高,引发速率增加。而聚丙烯酰胺凝胶反应为链式反应,即一旦被引发后将持续进行直至反应完成。所以,局部过高的自由基浓度将造成整个反应引发速率升高并持续进行到底。显然,悬浮液固相体积分数越高,黏度越大,反应引发效率越高,具体表现为反应诱导期越短(如图11(b)所示)。对于固相体积分数为20%的悬浮液而言,其凝胶反应的诱导期为340 s,而当固相体积分数增至50%时,其凝胶反应的诱导期缩短到190 s。此外,悬浮液的高黏度还会使得聚合反应进行缓慢,聚合速率下降,因此,凝胶反应的反应期延长(如图11(b)所示)。固相体积分数为50%的悬浮液,其反应期长达650 s,且在反应后期,反应体系的温度持续缓慢上升。对比图11(a)中各曲线还可知:随固相体积分数的增加,各“S”曲线中间部分的斜率逐渐降低,也表明凝胶反应的聚合速率逐渐下降;另外,反应体系的最高温度也随固相体积分数的增加而下降。当固相体积分数为20%时,反应体系的最高温度为36.6 ℃,而当固相体积分数为50%时,最高温度降低到30 ℃。固相体积分数的增加使凝胶反应体系的比热容也相应地增加,而聚合反应放出的能量一定,因此,反应体系的最高温度必然下降。
3.3 凝胶的均匀性
坯体结构均匀是凝胶注模的众多优点之一[5]。但如果凝胶注模过程控制不当,其优点就不能很好地体现出来,甚至有可能会成为缺点。显然,凝胶注模坯体为凝胶和粉体的复合体,因此,坯体结构的均匀性很大程度上受凝胶均匀性的影响,很有必要对凝胶的均匀性进行研究和讨论。
凝胶化学[18]认为凝胶形成过程是支化结构在整个体系内扩张并占据体系分摊整个空间的过程。但支化结构的发展并不是从体系的一点开始的,而是多个出发点同时开始生长并相互穿过边界而结合,最终链终止生长,获得凝胶物质。显然,凝胶的非均质性与支化结构的数量和分布有很大关系,而支化结构受凝胶反应条件如引发剂、催化剂、单体和交联剂等的浓度影响,特别与单体和交联剂的反应活性即竞聚率有关。凝胶反应条件影响凝胶反应速率和分子量即支化结构空间占有量。竞聚率不同使交联反应可能优先发生,或者最后发生,使交联点在聚合物中集中于某些特定部分,这些交联点密集的部位称为凝胶中的微凝胶。若这些微凝胶在宏观凝胶中分布不均匀,则成为非均质凝胶。在可见光波长范围内,由于光通过凝胶时产生散射,因而非均质凝胶是不透明的,而交联点均匀分布的凝胶,由于没有折射率的差别,因此,通常光照时是透明的。因此,可采用可见光分光光度计测试凝胶在某一波长的透射比来评价凝胶的均匀性。
凝胶的均匀性取决于其网络均匀性和结构均匀性,因此,从凝胶微观组织结构的角度来看,凝胶分为如下3种:均质网络均质结构凝胶、非均质网络非均质结构凝胶和均质网络非均质结构凝胶。图13所示为各种凝胶的网络结构示意图[18]。图13(a)所示的网络是交联点间分子量全部相等和交联点在空间分布均匀的均质网络链,称为“理想网络”,常作为理论模型使用。图13(b)所示的网络是由于交联点之间相对分子质量不同,交联空间分布不均匀的凝胶网络,实际上大多凝胶是这种结构。图13(c)的情况与图13(a)的一样,无分布、交联点间分子量相等,但是,交联点的空间位置不均匀,是非均质状态凝胶。
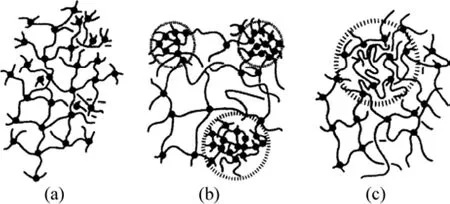
图13 3种不同凝胶的网络结构示意图[18]Fig.13 Schematic of three different gels network structure[18]
显然,通过控制凝胶反应条件如引发剂浓度、催化剂浓度、单体浓度、交联剂浓度和起始反应温度等,可获得具有网络较均匀且结构较均匀的凝胶,从而提高凝胶均匀性。网络均匀性主要是通过控制引发剂浓度和催化剂浓度等条件来实现。当引发剂和催化剂浓度较低时,凝胶反应引发速率较低,聚合速率也较小,单体的转化率较低,因此,聚合物的分子链长短不一,网络均匀性较差,凝胶均匀性不好。随引发剂和催化剂浓度的增加,反应引发速率和聚合速率逐渐增加,转化率升高,网络均匀性增加,凝胶的均匀性也增加。而当引发剂浓度较高时,反应引发速率和聚合速率急剧增加,此时,虽然单体转化率较高,但聚合反应所得产物的相对分子质量急剧下降,同时可能存在局部自由基浓度较高而出现“暴聚”的现象。所以,在引发剂和催化剂浓度较高条件下所得凝胶的网络结构均匀性下降,凝胶均匀性反而降低。但总体来说,引发剂和催化剂浓度对凝胶均匀性的影响有限。与单体浓度和m(AM)/m(MBAM)等反应条件相比较,引发剂和催化剂的加入只改变了凝胶反应速度,对最终凝胶微观组织的影响不大,相应地,对其均匀性的影响也不是很大(图2和图4)。
凝胶反应条件如单体浓度和交联剂浓度等不仅对凝胶网络均匀性,而且对结构均匀性有较大的影响。如前所述,单体浓度对聚合速率影响很大(图5)。当单体浓度较低时,引发速率过低,不利于单体聚合,所得凝胶的分子链不均匀,因此,其网络均匀性较差(图6);而当单体浓度较高时,其与交联剂进行交联形成支化结构的概率增加,因此结构均匀性变差,凝胶均匀性下降(图6)。此外,单体浓度对凝胶透明性的影响还可以归因于聚合热效应累积导致相分离,从而得到不均匀的混浊凝胶。因此,在凝胶注模的工艺过程中,须选择恰当的单体浓度,其较合适的范围(质量分数)为 10%~20%(图6)。交联剂浓度(单体交/联剂比例)对凝胶均匀性存在很大的影响(图8)。随交联剂浓度的增加即单体交/联剂比例的降低,其与单体进行交联形成的支化结构增加,所以凝胶的结构均匀性变差,凝胶均匀性下降。当交联剂浓度大于某一值时,所形成的水凝胶不透明。这可能是AM与MBAM在水中溶解度差别较大,随着m(AM)/m(MBAM)的降低,MBAM会聚集形成许多束状区域,这种聚集束的 MBAM 经聚合可形成高密度的聚合物,并达到可见光波长大小的链束,因而水凝胶透明性降低。这与 Gelfi等的研究[19]结果一致。此外,MBAM在水溶液中的溶解度恰好落在突变范围内。这进一步证实了凝胶透明性的变化是由 MBAM 溶解度的变化引起的。综上所述,为了获得具有均匀网络结构的透明凝胶,m(AM)/m(MBAM)应高于 20∶1(图8)。
起始反应温度对溶解度存在影响,所以,当起始反应温度较低时(如10 ℃),因交联剂MBAM在溶液中的溶解度较低而部分未溶,所以,合成的凝胶结构不均匀,呈乳白色。当起始反应温度较高时(如40 ℃),凝胶反应十分迅速,容易造成局部反应不均,凝胶网络均匀性下降,因此,凝胶透射比稍微降低。Gelfi等[17]研究温度对凝胶反应动力学的影响时发现:温度对凝胶均匀性存在很大影响,过低的温度甚至可使凝胶反应无法进行。因此,凝胶注模的起始反应温度也应控制在20~40℃(图10)。
4 结论
(1)引发剂浓度和催化剂浓度对凝胶反应速率有很大影响,但对凝胶均匀性的影响不大。随引发剂和催化剂浓度的增加,凝胶反应诱导期和反应期均迅速缩短。引发剂和催化剂浓度过高或过低均不利于凝胶反应的进行,影响凝胶微观组织结构的网络均匀性,因此,凝胶均匀性下降。可见,引发剂和催化剂浓度应控制适中。
(2)增加单体浓度,凝胶反应引发速率和聚合速率均相应地升高,因此,诱导期和反应期均缩短。单体浓度对凝胶均匀性有影响。当单体浓度较低时,所得凝胶的分子链不均匀,因此,其网络均匀性较差;当单体浓度较高时,其与交联剂形成的支化结构增加导致凝胶的结构均匀性变差,同时,聚合热效应累积增加导致相分离,所以,凝胶均匀性下降。故在凝胶注模的工艺过程中,单体质量分数较合适的范围为10%~20%。
(3)单体与交联剂质量比对凝胶反应以及诱导期和反应期影响不大,但对凝胶均匀性的影响很大。随着单体交与联剂质量比的降低,形成的支化结构增加,所以凝胶的结构均匀性变差,凝胶均匀性下降。当单体与交联剂质量比低于某一值时,由于交联剂溶解度较低而会聚集形成的束状区域在聚合过程中形成高密度的聚合物,因此凝胶均匀性降低。所以,对于凝胶注模的均匀性而言,单体与交联剂质量比应高于20∶1。
(4)起始反应温度对凝胶反应和均匀性都有很大影响。当起始反应温度较低时,因交联剂 MBAM 在溶液中的溶解度较低,凝胶结构不均匀;当起始反应温度较高时,过高的凝胶反应速度造成局部反应不均,形成的网络均匀性下降,凝胶均匀性降低。因此,凝胶注模的起始反应温度应控制在20~40 ℃。
(5)悬浮液固相体积分数的增加使得黏度升高,从而造成局部自由浓度过高,因此,能促进凝胶反应的进行,诱导期缩短。但悬浮液的高黏度还会使得聚合速率下降,因此,凝胶反应的反应期延长。
致谢:感谢天津大学的方道斌教授在实验过程中给予的帮助。
[1]Lange F F.Powder processing science and technology for increased reliability[J].Journal of the American Ceramic Society,1989,72(1): 3−15.
[2]Baader F H,Graule T J,Gauckler L J.Direct coagulation casting:A new green shaping technique.Part I: Processing principles[J].Industrial Ceramics,1996,16(1): 31−36.
[3]Lewis J A.Colloidal processing of ceramics [J].Journal of the American Ceramic Society,2000,83(10): 2341−2359.
[4]Olhero S M,Ganesh I,Torres P M C,et al.Aqueous colloidal processing of ZTA composites[J].Journal of the American Ceramic Society,2009,92(1): 9−16.
[5]王小锋,王日初,彭超群,等.凝胶注模成型技术的研究与进展[J].中国有色金属学报,2010,20(3): 496−509.WANG Xiao-feng,WANG Ri-chu,PENG Chao-qun,et al.Research and development of gelcasting[J].The Chinese Journal of Nonferrous Metals,2010,20(3): 496−509.
[6]Omatete O O,Janney M A,Strehlow R A.Gelcasting: A new ceramic forming process[J].American Ceramic Society Bulletin,1991,70(10): 1641−1649.
[7]Adolfsson E.Gelcasting of zirconia using agarose [J].Journal of the American Ceramic Society,2006,89(6): 1897−1902.
[8]DONG Man-jiang,MAO Xiao-jian,ZHANG Zhao-quan,et al.Gelcasting of SiC using epoxy resin as gel former[J].Ceramic International,2009,35(4): 1363−1366.
[9]GUO Dong,CAI Kai,LI Long-tu,et al.Gelcasting of PZT[J].Ceramics International,2003,29(4): 403−406.
[10]马利国,黄勇,杨金龙,等.凝胶注模成型固化过程及其影响因素: 陶瓷浆料凝胶点测定及其影响因素的研究[J].成都大学学报: 自然科学版,2002,21(2): 5−10.MA Li-guo,HUANG Yong,YANG Jin-long,et al.Solidification course and its influence factors for gelcasting: Gel point measurement of ceramics slurry and its influence factor[J].Journal of Chengdu University: Natural Science,2002,21(2):5−10.
[11]仝建峰,陈大明,李宝伟,等.氧化铝陶瓷凝胶注模成型凝固动力学研究[J].航空材料学报,2008,28(3): 49−52.TONG Jian-feng,CHEN Da-ming,LI Bao-wei,et al.Solidification kinetics of alumina suspension by gelcasting [J].Journal of Aeronautical Materials,2008,28(3): 49−52.
[12]戴春雷,杨金龙,黄勇.凝胶注模成型延迟固化研究[J].无机材料学报,2005,20(1): 83−89.DAI Chun-lei,YANG Jin-long,HUANG Yong.Investigation on delay solidification for gelcasting[J].Journal of Inorganic Materials,2005,20(1): 83−89.
[13]LI Fei,CHEN Hai-yan,WU Rui-zhi,et al.Effect of polyethylene glycol on the surface exfoliation of SiC green bodies prepared by gelcasting[J].Materials Science and Engineering A,2004,368: 255−259.
[14]方道斌,郭睿威,哈润华.丙烯酰胺聚合物[M].北京: 化学工业出版社,2006: 143−181.FANG Dao-bin,GUO Rui-wei,HA Run-hua.Polymers of acrylamide[M].Beijing: Chemical Industry Press,2006:143−181.
[15]张艳群,哈鸿飞.氯化钠相转变K-型卡拉胶/聚N-异丙基丙烯酰胺共混凝胶的辐射合成及性质研究[J].高分子学报,2001,33(4): 485−488.ZHANG Yan-qun,HA Hong-fei.Radiation synthesis and properties of Kappa-carrageenan-poly(N-isopropylacrylamide)hydrogel blends[J].Acta Polymerica Sinica,2001,33(4):485−488.
[16]潘祖仁.高分子化学[M].北京: 化学工业出版社,2006:78−80.PAN Zu-ren.Polymer Chemistry[M].Beijing: Chemical Industry Press,2006: 78−80.
[17]Gelfi C,Righetti P G.Polymerization kinetics of polyacrylamide gels II.Eeffect of temperature[J].Electrophoresis,1981,2(4):220−228.
[18]顾雪蓉,朱育平.凝胶化学[M].北京: 化学工业出版社,2004:57−117.GU Xue-rong,ZHU Yu-ping.Gel chemistry[M].Beijing:Chemical Industry Press,2004: 57−117.
[19]Gelfi C,Righetti P G.Polymerization kinetics of polyacrylamide gels I.Eeffect of different cross-linkers[J].Electrophoresis,1981,2(4): 213−219.
猜你喜欢
粘接(2021年2期)2021-06-10 01:08:11
云南化工(2020年11期)2021-01-14 00:51:00
含能材料(2020年8期)2020-08-10 06:44:20
选煤技术(2018年6期)2018-03-04 01:28:58
中国军转民(2017年7期)2017-12-19 13:30:00
大连工业大学学报(2015年4期)2015-12-11 04:06:50
石油化工(2015年9期)2015-08-15 00:43:05
橡塑技术与装备(2015年7期)2015-07-03 12:18:01
中国卫生(2014年10期)2014-11-12 13:10:24
中国神经精神疾病杂志(2014年1期)2014-03-01 03:23:23
