
含反式环己烷结构的C-葡萄糖苷类SGLT2抑制剂的合成及其降血糖活性Ⅱ
2012-11-21 02:15:02赵文静于秀玲王玉丽徐为人汤立达赵桂龙王建武
合成化学 2012年5期
赵文静, 于秀玲, 邵 华, 王玉丽, 徐为人, 汤立达, 赵桂龙,3, 王建武
(1. 山东大学 化学与化工学院,山东 济南 250100; 2. 天津药物研究院 药物设计与发现天津市重点实验室,天津 300193; 3. 山东轻工业学院 化学与制药工程学院,山东 济南 250353)
临床上具有不同作用机制的糖尿病治疗药物很多,如胰岛素类、二甲双胍类和磺酰脲类、胰岛素增敏剂、α-葡萄糖苷酶抑制剂以及新出现的DPP-Ⅳ抑制剂等[1]。尽管临床上治疗糖尿病的药物数量较多且作用机制也各不相同,但是由于传统降血糖药物具有明显的副作用,加之糖尿病具有不可逆转的特点,不同阶段需求不同作用机制的药物,因此临床上对糖尿病治疗药物仍然具有很大的需求。
钠-葡萄糖共转运体(SGLT)是一类主要在肾小管和肠道基底外侧膜表达的蛋白,其亚型主要有SGLT1和SGLT2两种,其中SGLT2主要分布在肾近端小管上,主要负责肾近端小管吸收原尿中的葡萄糖重新返回血液中[1,2]。大量试验表明,SGLT2抑制剂能有效抑制肾近端小管内SGLT2的活性而抑制原尿中葡萄糖的吸收,从而有效降低血糖浓度。天然产物根皮苷Phlorizin(Chart 1)是SGLT2的一种有效抑制剂,具有较强的诱导尿糖活性,但由于在肠道内容易被β-糖苷酶水解而不能被肠道吸收而无法口服[1]。早期制药工业以根皮苷的结构为蓝本,相继发现并报道了几种具有选择活性的SGLT2抑制剂,这些抑制剂多数是O-糖苷类(T-1095A, T-1095, Sergliflozin和Remogliflozin, Chart 1)。在开发自己的SGLT2抑制剂的过程中,我们也曾经设计、合成并研究了一些O-糖苷[3]、S-糖苷[4,5]和N-糖苷[6]。
在临床研究中发现,非C-糖苷类抑制剂,如O-糖苷、S-糖苷和N-糖苷等,其降血糖活性均比以Dapagliflozin(Chart 2)为代表的C-葡萄糖苷弱许多,且体内代谢稳定性也不理想,具有潜在的安全风险,后来均终止了临床试验。目前,部分仍然在临床上试验的SGLT2抑制剂(TS071, LX4211, Canagliflozin和ASP1941, Chart 2)均为C-糖苷类化合物。
基于上述情况,我们也设计、合成并研究了一些C-葡萄糖苷[7~10],其中包含了一类新型含反式环己烷结构的C-葡萄糖苷(Ⅰa~Ⅰd, Chart 2)[9],其分子右端含有碳原子依次增长的取代基(H, Me, Et和n-Pr)。体内生物活性测试发现,Ⅰa~Ⅰd均表现出较强的降血糖活性,且随着碳链的增长活性逐渐增加,在R=Et时(Ⅰc)达到顶端,R为n-Pr时(Ⅰd)略有下降。Ⅰd尽管活性不是最强,但是综合考虑其他生物学指标后,Ⅰd也进入了临床前开发阶段。随着Ⅰd进入临床前开发阶段,我们发现进一步拓展研究R为更长的烷烃基团显得非常必要,因为这对于我们深入了解这类化合物的完整全面的构效关系具有重要的意义。
本文设计并合成了一系列含有反式-4-烷基环己烷结构的C-葡萄糖苷类SGLT2抑制剂——反式-1-{4-甲氧基-3-[(4-取代环己基)甲基]苯基}-1-脱氧-β-D-吡喃葡萄糖(12a~12d, Scheme 1),其结构经1H NMR和MS表征。
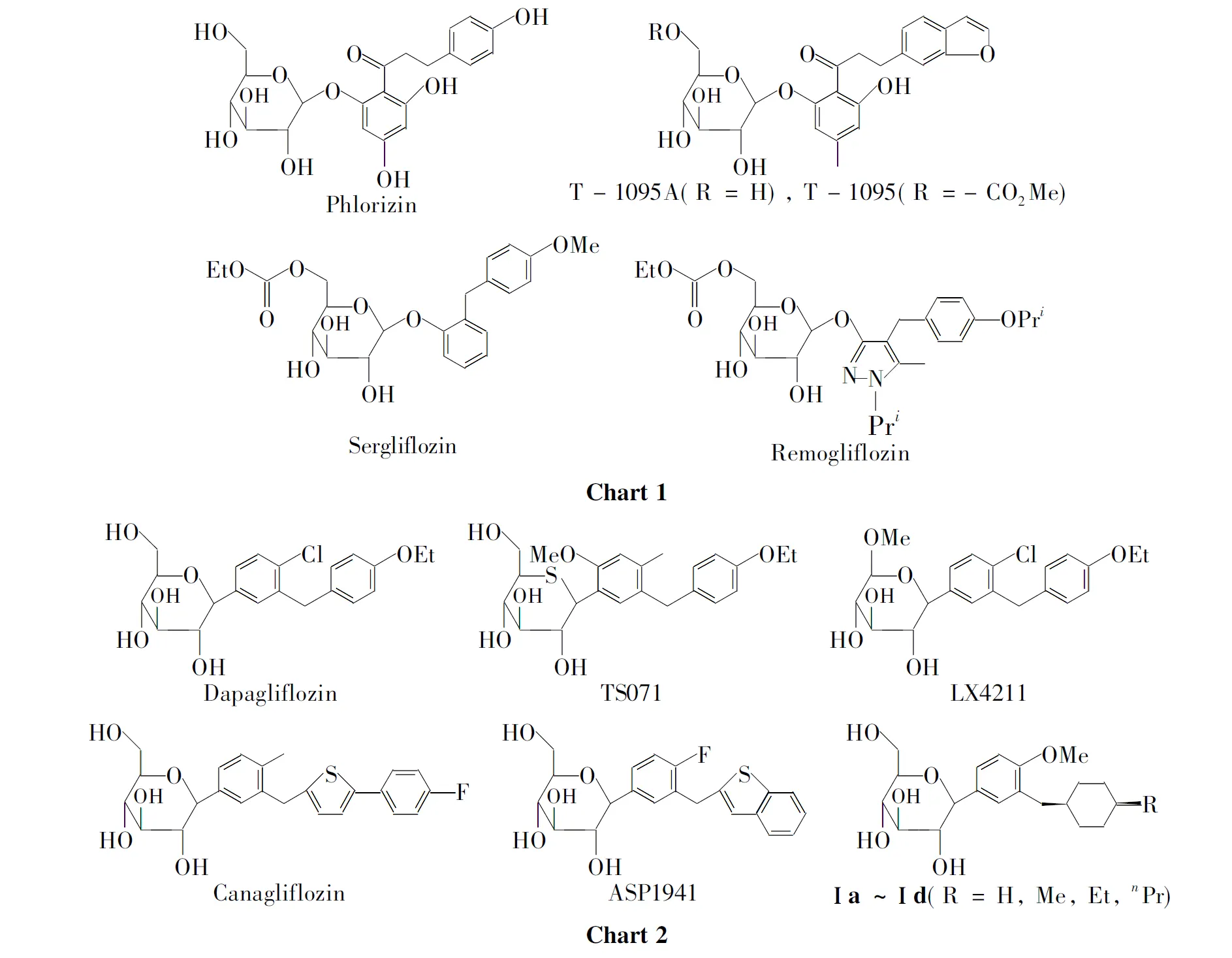
研究了R为更长的烷烃基团的化合物的合成,进一步完善了这类具有良好前景的SGLT2抑制剂的合成方法,进行了体内降血糖实验,得到了较为完整的构效关系。小鼠口服糖耐量实验表明,12a~12d均显示较强的降血糖活性。
1 实验部分
1.1 仪器与试剂
X-4型数字显微熔点仪(温度计未校正);Bruker AV400型核磁共振仪(DMSO-d6为溶剂,TMS为内标);Agilent Q-TOF 6510型高分辨质谱仪(ESI)。
D-葡萄糖酸内酯(6)按文献[2,12]方法转化为全三甲基硅烷基保护的化合物7; THF和甲苯用二苯甲酮作指示剂在金属钠存在下蒸馏作干燥处理;CH2Cl2和DMF分别在CaH2存在下常压和减压蒸馏作干燥处理;吡啶在块状固体KOH存在下蒸馏作干燥处理;甲醇用镁带干燥后蒸馏除水;草酰氯和乙酸酐在使用前新蒸馏;所有干燥后的溶剂均在氮气保护下室温密封保存并尽快使用。
1.2 合成
(1) (5-溴-2-羟基苯基)(反式-4-取代环己基)甲酮(4a~4d)的合成
在干燥的圆底烧瓶中依次加入反式-4-环己烷甲酸(1a~1d) 20 mmol和CH2Cl250 mL,搅拌下于室温缓慢滴加草酰氯25.39 g(30 mmol),滴毕,加2滴DMF,反应过夜。旋蒸脱溶和过量的草酰氯得无色液体2a~2d粗品。
2a~2d粗品用CH2Cl250 mL溶解,搅拌下加入4-溴苯甲醚2.44 g(20 mmol),冰水浴冷却下慢慢分批加入无水AlCl34.00 g(30 mmol),加毕,于室温反应过夜(TLC分析显示反应混合物中含有3a~3d,4a~4d和少量副产物3a′~3d′)。
在反应混合物中加入无水AlCl31.33 g(10 mmol),氮气氛中回流反应5 h(TLC分析显示反应混合物中含有4a~4d和少量副产物3a′~3d′,而3a~3d已经完全转化为4a~4d)。小心倾入冰水(200 mL)中,搅拌,分液,水相用CH2Cl2(100 mL)萃取,合并有机相,用饱和食盐水(2×100 mL)洗涤,无水硫酸钠干燥,旋蒸脱溶得油状液体4a~4d粗品,经柱层析[洗脱剂:A=V(乙酸乙酯) ∶V(石油醚)=1 ∶50]纯化得白色固体4a~4d。
4a: 产率89%, m.p.67 ℃~69 ℃;1H NMRδ: 11.73(bs, 1H), 7.93(d,J=2.4 Hz, 1H), 7.61(dd,J=2.8 Hz, 8.8 Hz, 1H), 6.93(d,J=8.8 Hz, 1H), 3.34~3.40(m, 1H), 1.84~1.87(m, 2H), 1.70~1.73(m, 2H), 1.38~1.44(m, 1H), 1.26~1.35(m, 2H), 1.08~1.18(m, 2H), 1.03~1.07(m, 1H), 0.85(d,J=6.8 Hz, 6H); HR-MSm/z: Calcd for C16H22O2Br{[M(79Br)+H]+} 325.080 3, found 325.080 5; for [M(81Br)+H]+327.078 3, found 327.078 8。
4b: 产率91%, m.p.89 ℃~90 ℃;1H NMRδ: 7.90(d,J=2.4 Hz, 1H), 7.61(dd,J=2.6 Hz, 9.0 Hz, 1H), 6.93(d,J=8.8 Hz, 1H), 3.35~3.41(m, 1H), 1.75~1.84(m, 4H), 1.18~1.37(m, 9H), 1.02~1.08(m, 2H), 0.86(t,J=6.8 Hz, 3H); HR-MSm/z: Calcd for C17H24O2Br{[M(79Br)+H]+} 339.096 0, found 339.095 3; for [M(81Br)+H]+341.093 9, found 341.093 4。
4c: 产率87%, m.p.95 ℃~96 ℃;1H NMRδ: 11.75(bs, 1H), 7.95(d,J=2.4 Hz, 1H), 7.62(dd,J=2.4 Hz, 8.8 Hz, 1H), 6.93(d,J=9.2 Hz, 1H), 3.34~3.41(m, 1H), 1.87~1.90(m, 2H), 1.78~1.81(m, 2H), 1.25~1.35(m, 2H), 1.10~1.20(m, 2H), 0.95~1.02(m, 1H), 0.84(s, 9H); HR-MSm/z: Calcd for C17H24O2Br{[M(79Br)+H]+} 339.096 0, found 339.095 8; for [M(81Br)+H]+341.093 9, found 341.093 7。
4d: 产率85%, m.p.90 ℃~91 ℃;1H NMRδ: 11.67(bs, 1H), 7.90(d,J=2.4 Hz, 1H), 7.61(dd,J=2.6 Hz, 9.0 Hz, 1H), 6.93(d,J=8.8 Hz, 1H), 3.34~3.42(m, 1H), 1.75~1.84(m, 4H), 1.16~1.37(m, 11H), 1.00~1.08(m, 2H), 0.85(t,J=7.0 Hz, 3H); HR-MSm/z: Calcd for C18H26O2Br{[M(79Br)+H]+} 353.111 6, found 353.112 0; for [M(81Br)+H]+355.109 6, 355.110 0。
(2) (5-溴-2-甲氧基苯基)(反式-4-取代环己基)甲酮(3a~3d)的合成
在干燥的圆底烧瓶中依次加入4a~4d16 mmol的DMF(40 mL)溶液, MeI 2.84 g(20 mmol)和固体K2CO34.15 g(30 mmol),搅拌下于室温反应过夜(TLC分析显示4a~4d完全转化为3a~3d)。倾入冰水(300 mL)中,搅拌下用浓盐酸小心调至pH 4~5,用CH2Cl2(3×100 mL)萃取,合并萃取液,用饱和食盐水(2×100 mL)洗涤,无水硫酸钠干燥,旋蒸脱溶得油状液体3a~3d粗品,经硅胶柱层析(洗脱剂: A=1 ∶30)纯化得无色油状液体3a~3d。
3a: 产率96%;1H NMRδ: 7.64(dd,J=2.4 Hz, 8.8 Hz, 1H), 7.48(d,J=2.4 Hz, 1H), 7.12(d,J=8.8 Hz, 1H), 3.84(s, 3H), 2.96~3.03(m, 1H), 1.83~1.86(m, 2H), 1.71~1.73(m, 2H), 1.37~1.41(m, 1H), 1.16~1.26(m, 2H), 0.97~1.02(m, 3H), 0.83(d,J=6.8 Hz, 6H); HR-MSm/z: Calcd for C17H24O2Br{[M(79Br)+H]+} 339.096 0, found 339.096 3; for [M(81Br)+H]+341.093 9, found 341.094 4。
3b: 产率95%;1H NMRδ: 7.64(dd,J=2.6 Hz, 9.0 Hz, 1H), 7.48(d,J=2.8 Hz, 1H), 7.12(d,J=8.8 Hz, 1H), 3.84(s, 3H), 2.97~3.04(m, 1H), 1.74~1.83(m, 4H), 1.20~1.26(m, 6H), 1.15~1.16(m, 3H), 0.90~0.96 (m, 2H), 0.85(t,J=7.0 Hz, 3H); HR-MSm/z: Calcd for C18H26O2Br{[M(79Br)+H]+} 353.111 6, found 353.111 8; for [M(81Br)+H]+355.109 6, found 355.109 9。
3c: 产率97%;1H NMRδ: 7.64(dd,J=2.8 Hz, 9.2 Hz, 1H), 7.49(d,J=2.8 Hz, 1H), 7.12(d,J=8.8 Hz, 1H), 3.84(s, 3H), 2.95~3.01(m, 1H), 1.86~1.89(m, 2H), 1.77~1.79(m, 2H), 1.15~1.25(m, 2H), 0.94~1.05(m, 3H), 0.82(s. 9H); HR-MSm/z: Calcd for C18H26O2Br{[M(79Br)+H]+} 353.111 6, found 353.112 1; for [M(81Br)+H]+355.109 6, found 355.110 0。
3d: 产率94%;1H NMRδ: 7.64(dd,J=2.8 Hz, 8.8 Hz, 1H), 7.49(d,J=2.8 Hz, 1H), 7.12(d,J=9.2 Hz, 1H), 3.84(s, 3H), 2.98~3.04(m, 1H), 1.74~1.83(m, 4H), 1.20~1.29(m, 8H), 1.14~1.19(m, 3H), 0.85(t,J=7.0 Hz, 3H); HR-MSm/z: Calcd for C19H28O2Br{[M(79Br)+H]+} 367.127 3, found 367.127 3; for [M(81Br)+H]+369.125 2, found 369.125 1。
(3) 4-溴-2-[(反式-4-取代环己基)甲基]苯甲醚(5a~5d)的合成
在干燥圆底烧瓶中加入3a~3d15 mmol的CH2Cl2(20 mL)溶液,搅拌下于室温加入Et3SiH 6.98 g(60 mmol),加毕,慢慢滴加BF3·Et2O 4.26 g(30 mmol),滴毕,反应过夜(TLC跟踪)。慢慢加入饱和NaHCO3溶液100 mL,于室温搅拌30 min;加入CH2Cl2100 mL和饱和食盐水200 mL,振荡,分液,水相用CH2Cl2(100 mL)萃取,合并有机相,用饱和食盐水(100 mL)洗涤,无水硫酸钠干燥,旋蒸脱溶得油状液体5a~5d粗品,经硅胶柱层析(洗脱剂:A=1 ∶50)纯化得无色油状液体5a~5d。
5a: 产率90%;1H NMRδ: 7.31(dd,J=1.2 Hz, 8.8 Hz, 1H), 7.21(s, 1H), 6.89(d,J=8.8 Hz, 1H), 3.74(s, 3H), 2.40(d,J=6.8 Hz, 2H), 1.58~1.62(m, 4H), 1.33~1.40(m, 2H), 0.84~0.94(m, 5H), 0.81(d,J=6.4 Hz, 6H); HR-MSm/z: Calcd for C17H26OBr{[M(79Br)+H]+} 325.116 7, found 325.116 6; for [M(81Br)+H]+327.114 7, found 327.114 4。
5b: 产率91%;1H NMRδ: 7.31(dd,J=2.4 Hz, 8.8 Hz, 1H), 7.21(d,J=2.4 Hz, 1H), 6.89(d,J=8.8 Hz, 1H), 3.74(s, 3H), 2.40(d,J=7.2 Hz, 2H), 1.64~1.67(m, 2H), 1.54~1.57(m, 2H), 1.38~1.44(m, 1H), 1.22~1.23(m, 4H), 1.11(s, 3H), 0.73~0.97(m, 7H); HR-MSm/z: Calcd for C18H28OBr{[M(79Br)+H]+} 339.132 4, found 339.132 2; for [M(81Br)+H]+341.130 3, found 341.130 0。
5c: 产率94%;1H NMRδ: 7.31(dd,J=2.4 Hz, 8.8 Hz, 1H), 7.21(d,J=2.4 Hz, 1H), 6.89(d,J=8.8 Hz, 1H), 3.74(s, 3H), 2.40(d,J=7.2 Hz, 2H), 1.61~1.68(m, 4H), 1.39(m, 1H), 0.84~0.94(m, 5H), 0.79(s, 9H); HR-MSm/z: Calcd for C18H28OBr{[M(79Br)+H]+} 339.132 4, found 339.132 3; for [M(81Br)+H]+341.130 3, found 341.130 4。
5d: 产率90%;1H NMRδ: 7.31(dd,J=2.6 Hz, 8.6 Hz, 1H), 7.21(d,J=2.8 Hz, 1H), 6.89(d,J=8.8 Hz, 1H), 3.74(s, 3H), 2.40(d,J=6.8 Hz, 2H), 1.64~1.67(m, 2H), 1.54~1.57(m, 2H), 1.39~1.43(m, 1H), 1.16~1.27(m, 6H), 1.09~1.10(m, 3H), 0.75~0.96(m, 7H); HR-MSm/z: Calcd for C19H30OBr{[M(79Br)+H]+} 353.148 0, found 353.148 5; for [M(81Br)+H]+355.146 0, found 355.146 3。
(4) 反式-1-{4-甲氧基-3-[(4-烷基环己基)甲基]苯基}-α/β-D-甲基吡喃葡萄糖苷(8a~8d)的合成
在干燥的圆底烧瓶中加入5a~5d10 mmol的THF(20 mL)溶液和一枚干燥磁子,以干燥的氮气吹扫后以橡胶软塞封口。将其置于盛有液氮/乙醇的Dewar瓶中冷却至-78 ℃,启动电磁搅拌,用注射器慢慢滴加n-BuLi 12 mmol的正己烷溶液7.5 mL,滴毕,反应30 min;用注射器慢慢滴加7 7.00 g(15 mmol)的甲苯(10 mL)溶液[2,12],滴毕,反应30 min;升至室温反应60 min。小心倾入冰水(200 mL)中,搅拌下用浓盐酸调至pH 2~3,搅拌15 min;用CH2Cl2(2×50 mL)萃取,合并萃取液,用饱和食盐水洗涤,无水硫酸钠干燥,旋蒸脱溶得深黄色油状液体8a~8d粗品(不纯化,直接用于下一步反应)。
(5) 反式-1-{4-甲氧基-3-[(4-烷基环己基)甲基]苯基}-1-脱氧-α/β-D-吡喃葡萄糖(9a~9d)的合成
在干燥的圆底烧瓶中加入8a~8d粗品的CH2Cl2(20 mL)溶液,搅拌下于-30 ℃加入Et3SiH 2.33 g(20 mmol),缓慢滴加BF3·Et2O 1.42 g (10 mmol)的CH2Cl2(5 mL)溶液,滴毕,于室温反应过夜。加入饱和NaHCO3溶液20 mL搅拌30 min,用5%盐酸调至pH 3~4,加入饱和食盐水50 mL和CH2Cl250 mL,搅拌5 min后静置分层,水相用CH2Cl2(50 mL)萃取,合并有机相,用饱和食盐水洗涤,无水硫酸钠干燥,旋蒸脱溶得褐色油状液体9a~9d粗品(不纯化,直接用于下一步反应)。
(6) 反式-2,3,4,6-四-O-乙酰基-1-{4-甲氧基-3-[(4-烷基环己基)甲基]苯基}-1-脱氧-α/β-D-吡喃葡萄糖(11a~11d)的合成
在圆底烧瓶中加入9a~9d粗品的吡啶(30 mL)溶液,冰水浴冷却,搅拌下慢慢滴加乙酸酐10.21 g(100 mmol),滴毕,加入DMAP(4-二甲氨基吡啶)0.50 g,于室温反应过夜。倾入冰水(300 mL)中,用CH2Cl2(3×50 mL)萃取,合并萃取液,依次用1%盐酸和饱和食盐水洗涤,无水硫酸钠干燥,旋蒸脱溶得褐色油状液体10a~10d粗品,经柱层析(洗脱剂:A=1 ∶3)纯化,用混合溶剂[V(乙酸乙酯) ∶V(石油醚)=1 ∶5]重结晶得白色固体11a~11d。
11a: 产率72%, m.p.156 ℃~157 ℃;1H NMRδ: 7.14(dd,J=2.0 Hz, 8.4 Hz, 1H), 7.00(d,J=2.0 Hz, 1H), 6.90(d,J=8.4 Hz, 1H), 5.33(t,J=9.4 Hz, 1H), 5.06(t,J=9.6 Hz, 1H), 5.00(t,J=9.8 Hz, 1H), 4.57(d,J=10.0 Hz, 1H), 4.02~4.14(m, 3H), 3.74(s, 3H), 2.35~2.45(m, 2H), 2.02(s, 3H), 2.00(s, 3H), 1.93(s, 3H), 1.73(s, 3H), 1.57~1.61(m, 4H), 1.32~1.40(m, 2H), 0.86~0.97(m, 5H), 0.81(d,J=6.8 Hz, 6H); HR-MSm/z: Calcd for C31H45O10{[M+H]+} 577.301 3, found 577.301 1。
11b: 产率70%, m.p.153 ℃~254 ℃;1H NMRδ: 7.14(dd,J=2.0 Hz, 8.4 Hz, 1H), 6.99(d,J=2.0 Hz, 1H), 6.89(d,J=8.4 Hz, 1H), 5.32(t,J=9.6 Hz, 1H), 5.05(t,J=9.6 Hz, 1H), 5.00(t,J=9.6 Hz, 1H), 4.57(d,J=10.0 Hz, 1H), 4.01~4.12(m, 3H), 3.74(s, 3H), 2.38~2.41(m, 2H), 2.01(s, 3H), 1.99(s, 3H), 1.92(s, 3H), 1.71(s, 3H), 1.63~1.65(m, 2H), 1.51~1.56(m, 2H), 1.39~1.41(m, 1H), 1.21~1.22(m, 4H), 1.11(s, 3H), 0.85~0.91(m, 2H), 0.84(t,J=6.8 Hz, 3H), 0.77~0.78(m, 2H); HR-MSm/z: Calcd for C32H47O10{[M+H]+} 591.316 9, found 591.317 3。
11c: 产率76%, m.p.129 ℃~130 ℃;1H NMRδ: 7.14(dd,J=2.0 Hz, 8.4 Hz, 1H), 7.00(d,J=2.0 Hz, 1H), 6.90(d,J=8.4 Hz, 1H), 5.33(t,J=9.6 Hz, 1H), 5.06(t,J=9.6 Hz, 1H), 5.01(t,J=9.8 Hz, 1H), 4.58(d,J=10.0 Hz, 1H), 4.08~4.13(m, 2H), 4.03~4.06(m, 1H), 3.75(s, 3H), 2.38~2.42(m, 2H), 2.02(s, 3H), 2.00(s, 3H), 1.93(s, 3H), 1.73(s, 3H), 1.61~1.68(m, 4H), 1.40(s, 1H), 0.86~0.88(m, 4H), 0.80(s, 9H); HR-MSm/z: Calcd for C32H47O10{[M+H]+} 591.316 9, found 591.317 1。
11d: 产率77%, m.p.145 ℃~146 ℃;1H NMRδ: 7.14(dd,J=2.0 Hz, 8.4 Hz, 1H), 6.99(d,J=2.0 Hz, 1H), 6.89(d,J=8.8 Hz, 1H), 5.32(t,J=9.4 Hz, 1H), 5.05(t,J=9.6 Hz, 1H), 5.00(t,J=9.6 Hz, 1H), 4.57(d,J=10.0 Hz, 1H), 4.01~4.13(m, 3H), 3.73(s, 3H), 2.40(dd,J=2.8 Hz, 6.8 Hz, 1H), 2.01(s, 3H), 1.99(s, 3H), 1.92(s, 3H), 1.71(s, 3H), 1.64~1.65(m, 2H), 1.51~1.54(m, 2H), 1.38~1.41(m, 1H), 1.17~1.27(m, 6H), 1.10~1.15(m, 3H), 0.88~0.94(m, 2H), 0.84(t,J=6.8 Hz, 3H), 0.71~0.78(m, 2H); HR-MSm/z: Calcd for C33H49O10{[M+H]+} 605.332 6, found 605.332 7。
(7)12a~12d的合成
在干燥圆底烧瓶中加入无水甲醇10 mL和金属钠46 mg(2 mmol),搅拌下于室温反应至所有金属钠消失。加入11a~11d5 mmol,于室温反应至终点(一般在1 h以内)。加入干燥的强酸性阳离子交换树脂(H+型)2.0 g,于室温搅拌过夜(溶液pH 7)。抽滤,滤饼用甲醇(20 mL)洗涤,洗涤液与滤液合并,旋蒸除去溶剂,残余物于室温真空干燥得白色泡沫12a~12d。
12a: 产率99%;1H NMRδ: 7.11(d,J=8.4 Hz, 1H), 7.01(s, 1H), 6.86(d,J=8.0 Hz, 1H), 4.87~4.88(m, 2H), 4.63(d,J=5.6 Hz, 1H), 4.40(t,J=5.6 Hz, 1H), 3.91(d,J=9.2 Hz, 1H), 3.80(s, 3H), 3.67~3.71(m, 1H), 3.40~3.46(m, 1H), 3.12~3.28(m, 4H), 2.47(dd,J=6.4 Hz, 12.8 Hz, 1H), 2.34(dd,J=7.2 Hz, 12.8 Hz, 1H), 1.62~1.64(m, 4H), 1.33~1.39(m, 2H), 0.89~0.97(m, 5H), 0.81(d,J=6.8 Hz, 6H); HR-MSm/z: Calcd for C23H36O6Na{[M+Na]+} 431.241 0, found 431.241 2。
12b: 产率100%;1H NMRδ: 7.11(dd,J=2.0 Hz, 8.4 Hz, 1H), 7.01(d,J=2.0 Hz, 1H), 6.86(d,J=8.8 Hz, 1H), 4.40(bs, 4H), 3.91(d,J=9.2 Hz, 1H), 3.74(s, 3H), 3.68~3.71~(m, 1H), 3.43(dd,J=1.6 Hz, 11.6 Hz, 1H), 3.12~3.27(m, 4H), 2.44~2.47(m, 1H), 2.31~2.37(m, 1H), 1.62~1.68(m, 4H), 1.39~1.43(m, 1H), 1.23~1.25(m, 4H), 1.13(s, 3H), 0.79~0.98(m, 7H); HR-MSm/z: Calcd for C24H38O6Na{[M+Na]+} 445.256 6, found 445.256 5。
12c: 产率98%;1H NMRδ: 7.10~7.12(m, 1H), 7.01(d,J=1.6 Hz, 1H), 6.86(d,J=8.8 Hz, 1H), 4.86~4.88(m, 2H), 4.63(d,J=6.4 Hz, 1H), 4.40(t,J=5.6 Hz, 1H), 3.91(d,J=9.2 Hz, 1H), 3.74(s, 3H), 3.67~3.71(m, 1H), 3.40~3.45(m, 1H), 3.22~3.28(m, 1H), 3.12~3.19(m, 3H), 2.44~2.47(m, 1H), 2.31~2.36(m, 1H), 1.68~1.70(m, 4H), 1.37~1.42(s, 1H), 0.91~0.96(m, 5H), 0.80(s, 9H); HR-MSm/z: Calcd for C24H38O6Na {[M+Na]+} 445.256 6, found 445.256 4。
12d: 产率99%;1H NMRδ: 7.10(dd,J=1.8 Hz, 8.2 Hz, 1H), 7.00(d,J=1.6 Hz, 1H), 6.85(d,J=8.4 Hz, 1H), 4.85~4.87(m, 2H), 4.62(d,J=5.6 Hz, 1H), 4.39(t,J=5.8 Hz, 1H), 3.90(d,J=9.6 Hz, 1H), 3.73(s, 3H), 3.66~3.70(m, 1H), 3.40~3.45(m, 1H), 3.11~3.27(m, 4H), 2.43~2.46(m, 1H), 2.34(dd,J=7.2 Hz, 12.8 Hz, 1H), 1.61~1.67(m, 4H), 1.40(s, 1H), 1.19~1.28(m, 6H), 1.09~1.11(m, 3H), 0.88~0.97(m, 2H), 0.84(t,J=6.8 Hz, 3H), 0.78(s, 2H); HR-MSm/z: Calcd for C25H40O6Na{[M+Na]+} 459.272 3, found 459.272 0。
2 结果与讨论
2.1 合成
DMF催化1a~1d与(COCl)2在CH2Cl2中于室温反应制得对应的酰氯2a~2d。 AlCl3催化2a~2d粗品与4-溴苯甲醚在CH2Cl2中发生Friedel-Crafts酰基化反应,区域选择性地在4-溴苯甲醚分子OMe的邻位引入酰基得到3a~3d,没有观察到因在4-溴苯甲醚分子Br原子的邻位引入酰基而生成的潜在区域异构体。在AlCl3存在下3a~3d在CH2Cl2中回流脱去甲基得到对应的酚4a~4d,其作用机制可能涉及3a~3d分子中的C=O和MeO与AlCl3分子由配位作用而形成的稳定六元环有关[9,11~13]。4a~4d经柱层析和重结晶纯化后得到的纯品使用MeI/K2CO3在DMF中于室温顺利甲基化得到预期产物3a~3d。
合成3a~3d的路线源于对文献[9]方法的改进(Scheme 2)。酰氯2a~2d与4-溴苯甲醚在无水AlCl3的催化下发生Friedel-Crafts酰基化反应,得到了预期产物3a~3d,同时观察到一个副产物,经过结构鉴定,发现为脱溴化合物3a′~3d′。由于反应体系中AlCl3的存在,生成的3a~3d会发生脱甲基化反应得到4a~4d。
前期研究[9]发现该脱甲基化反应在室温下即可完成,但是进一步的研究发现,该反应对温度较为敏感,在室温较高时(如夏天),于室温反应即可进行得比较完全;但是在室温较低时(如春秋冬),在室温下也可发生反应且难以避免,但是即使在大幅度延长反应时间的情况下也难以进行完全;尝试进一步降低温度来避免该反应,结果未能如愿。因为低温往往导致Friedel-Crafts酰基化反应(2→3)的速度大大降低甚至反应不完全而影响收率。为了得到3→4的可靠反应条件,我们在Friedel-Crafts酰基化反应完成后在反应混合物中补加一定量的无水AlCl3并升温回流,此时3a~3d即可完全脱去甲基转化为4a~4d。需要指出的是,此时副产物3a′~3d′并未脱去甲基,因为其分子中的C=O与MeO处于苯环的对位,不能象3a~3d那样与AlCl3形成稳定的分子内六元环[9,11~13]。
Friedel-Crafts酰基化反应完成后经过处理,得到了4a~4d和3a′~3d′的混合物,我们[9]曾将该混合物粗品直接甲基化后得到3a~3d和3a′~3d′的混合物,再经柱层析分离。但3a~3d和3a′~3d′的极性非常相近,因此导致通过柱层析分离非常困难,尤其在放大实验中更加明显。
本文经过优化改进,先用柱层析分离4a~4d与3a′~3d′,因为两者极性相差较大,所以通过柱层析分离比较容易。纯品4a~4d甲基化后得到3a~3d。也曾用碱水溶液洗涤的方法分离4a~4d与3a′~3d′,结果并不理想。因为4a~4d分子中疏水性基团较大,其盐的水溶性较差,而且由于C=O导致的邻位CH的酸性较大的原因4a~4d分子中的反式环己烷结构经过强碱处理后会发生显著的异构化,生成大量的顺式异构体4a′~4d′(Scheme 3),给分离带来很大的困难;但是这种异构化在甲基化反应条件下,即使存在K2CO3也不会发生。
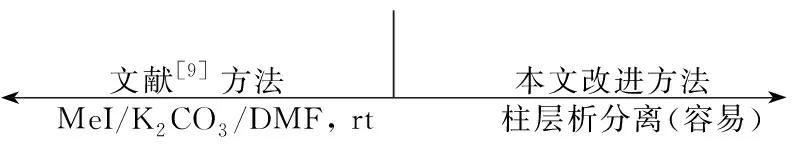

Scheme2


Scheme3
羰基化合物3a~3d使用Et3SiH/BF3·Et2O在CH2Cl2中于室温还原,得到对应的亚甲基化合物5a~5d。D-葡萄糖酸内酯(6)使用已知的方法转化为其全三甲基硅烷基保护的化合物7[2,12]。溴化物5a~5d在低温下使用n-BuLi处理,得到溴-锂交换产物芳基锂,后者与7发生亲核加成反应,生成游离半缩酮8a~8d。很明显8a~8d是异头体混合物,不需要进一步纯化和表征而直接用于下一步还原反应。需要指出的是,在我们前期的研究[9]中,8a~8d先用MeOH/MsOH室温下处理而发生甲基糖苷化将其转化为其对应的甲基糖苷再进行还原,经过优化研究,我们发现该步酸催化的甲基糖苷化没有必要,8a~8d可以直接还原。8a~8d在CH2Cl2中使用Et3SiH/BF3·Et2O于-30 ℃还原,顺利得到脱去异头位OH的C-葡萄糖苷9a~9d。
本文改进了此步的反应温度,由我们前期研究[9]的室温改为此处优化后的-30 ℃,因为我们发现该还原反应在低温下即非常容易发生,而且低温下可以提高9a~9d的β/α比例,在室温下还原β/α接近9/1,而在-30 ℃下还原可以提高到>99/1。9a~9d是β/α的异头物混合物,其中期望的β式占主要比例,直接分离9a~9d中β/α两个异构体比较困难,因此我们将9a~9d用传统的Ac2O/吡啶在DMAP(4-二甲氨基吡啶)的催化下在室温下顺利进行全乙酰化而得到极性较小的10a~10d,此步反应经过优化后避免了使用原先研究中[9]条件较为剧烈的“AcONa/Ac2O/AcOH/回流”的条件,大大简化了操作。10a~10d经过柱层析和结晶后分离出纯品β-异头体11a~11d;再于室温用MeONa/MeOH处理脱去所有乙酰基得目标化合物12a~12d。
2.2 体内降糖活性
化合物的降血糖活性测试采用小鼠口服葡萄糖耐受量实验(OGTT)评价[7~9]。
样品用1%羧甲基纤维素钠水溶液配制成1 mg·mL-1的混悬液,给药容量为0.4 mL·(20 g体质量)-1,相当于20 mg·kg-1的剂量。健康ICR小鼠,雌雄各半,体质量20 g~24 g,符合一级标准。
实验小鼠禁食16 h,分别给小鼠口服待测化合物或空白1%羧甲基纤维素钠水溶液,90 min后腹腔注射2 g·kg-1的葡萄糖生理盐水溶液,2 h后再次注射葡萄糖,空白组不给糖。于初次给糖后0.25 h, 0.5 h, 1.0 h, 1.5 h, 2.0 h, 2.5 h, 3.0 h, 3.5 h和4 h分别用毛细管从小鼠眼球后静脉丛取血,然后离心分离血清,采用葡萄糖氧化酶法测定各时间点血清中葡萄糖的含量。
化合物的降血糖活性以血糖抑制率(%)衡量,测试结果见表1。
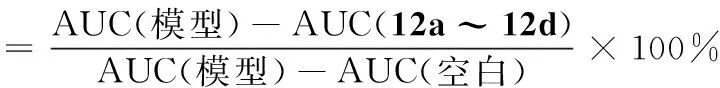
式中:AUC为“血糖浓度-时间”曲线的曲线下面积
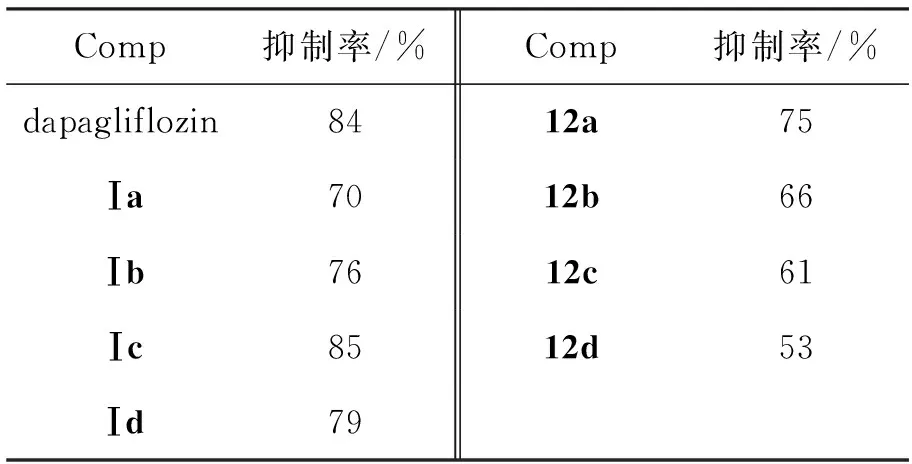
表 1 化合物的OGTT血糖抑制率*Table 1 Blood sugar inhibitory rates of compounds in OGTT
*给药量20 mg·kg-1, Ⅰa~Ⅰd按文献[9]方法制备
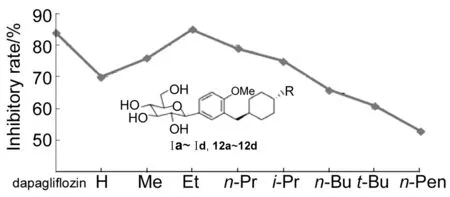
R图 1 含反式-4-烷基环己烷C-葡萄糖苷的构效关系Figure 1 SAR of trans-4-alkylcyclohexane-bearing C-glucosides
为了比较Ⅰa~Ⅰd[9]与12a~12d的降血糖活性,根据表1数据,以取代基R为横坐标以血糖抑制率为纵坐标作图,得到构效关系(SAR)图(图1)。由图1可见,取代基R从左到右依次增加碳原子的个数,对于丙基和丁基这两个基团,各有两个同碳数目的异构体,其排列从左到右直链在前支链在后。随着取代基R从H到正戊基的依次变化,抑制率呈现出先增加后减小的有规律的趋势,在R=Et时(Ⅰc)达到最大值(85%),此时已经超过阳性对照化合物dapagliflozin(84%)。Ⅰc和Ⅰd的抑制率均较高,而在R中含有的碳原子数超过4个(R=n-Bu,t-Bu和n-Pentyl)后,抑制率迅速下降。对比Chart 1和Chart 2中所列的公认的SGLT2抑制剂可以看出,它们的分子右端苯环上的侧链长度多为2个~3个非氢原子(如OMe和OEt),因此本研究系列所得到的结论与之一致;同时也说明,SGLT2抑制剂的分子右端也可以容忍非芳香性环状结构。上述结论对SGLT2抑制剂的设计和合成具有重要的指导意义。
[1] Washburn W N. Development of the renal glucose reabsorption inhibitors:A new mechanism for the pharmacotherapy of diabetes mellitus type 2[J].J Med Chem,2009,52:1785-1794.
[2] Meng M, Ellsworth B A, Nirschl A A,etal. Discovery of dapagliflozin:A potent,selective renal sodium-dependent glucose cotransporter 2(SGLT2) inhibitor for the treatment of type 2 diabetes[J].J Med Chem,2008,51:1145-1149.
[3] 王致峰,赵桂龙,王玉丽,等. 苯萘基糖苷类化合物的合成及其治疗糖尿病的活性[J].合成化学,2009,17:570-573.
[4] Gao Y L, Zhao G L, Liu W,etal. Thiadiazole-based thioglycosides as sodium-glucose co-transporter 2(SGLT2) inhibitors[J].Chin J Chem,2010,28:605-612.
[5] 王致峰,赵桂龙,刘巍,等. 含噻二唑环的硫代糖苷类SGLT2 抑制剂的设计、合成与降血糖活性研究[J].有机化学,2010,30:849-859.
[6] Gao Y L, Zhao G L, Liu W,etal. Design, synthesis and in vivo hypoglycemic activity of tetrazole-bearingN-glycosides as SGLT2 inhibitors[J].Indian J Chem,2010,49B:1499-1508.
[7] 史永恒,赵桂龙,刘巍,等. 钠-葡萄糖协同转运蛋白抑制剂的合成及降血糖活性[J].中国药物化学杂志,2011,21:57-59.
[8] Shi Y H, Zhao G L, Lou Y Y,etal. gem-Dimethyl-bearingC-glucosides as sodium-glucose co-transporter 2(SGLT2) inhibitors[J].Chin J Chem,2011,29:1192-1198.
[9] 邵华,高云龙,楼袁媛,等. 含反式环己烷结构的C-葡萄糖苷类SGLT2抑制剂的设计、合成与降血糖活性研究[J].有机化学,2011,31:836-842.
[10] Zhao W J, Shi Y H, Zhao G L,etal. Design,synthesis and in vivo biological activity of gem-dimethyl-bearingC-glucosides as SGLT2 inhibitors[J].Chin Chem Lett,2011,22:1215-1218.
[11] Parker K A, Petraitis J J. Synthesis of ansamycins:An approach to the naphthoquinone portion of the rifamycins and streptovaricins[J].Tetrahedron Lett,1981,22:397-400.
[12] Li T T, Wu L Y. Facile regio- and stereoselective total synthesis of racemic aklavinone[J].J Am Chem Soc,1981,103:7007-7009.
[13] Kawamura Y, Takatsuki H, Torii F,etal. Studies of the selectiveO-alkylation and dealkylation of flavonoids. ⅩⅥ.Demethylation of 2′-methoxyacetophenones with anhydrous aluminum chloride or bromide in acetonitrile[J].Bull Chem Soc Jpn,1994,67:511-515.
[14] 邵华,赵桂龙,刘巍,等. SGLT2抑制剂Dapagliflozin的全合成[J].合成化学,2010,18:389-392.
猜你喜欢
中国生殖健康(2020年5期)2021-01-18 03:00:06
吉林农业(2019年6期)2019-06-11 03:10:30
中国生殖健康(2018年5期)2018-11-06 07:15:56
食品与机械(2018年5期)2018-07-16 01:34:00
食品与机械(2018年5期)2018-07-14 03:15:24
中成药(2017年10期)2017-11-16 00:50:15
中成药(2017年4期)2017-05-17 06:09:46
中国民族医药杂志(2016年5期)2016-05-09 07:43:50
橡胶工业(2016年2期)2016-02-23 21:36:51
医学美学美容·中旬刊(2015年2期)2015-10-21 19:58:27
