
高比表面积氟化镁的合成及其在催化中的应用研究进展
2012-10-19 03:35:52牛怀成李利春韩文峰唐浩东刘化章
化工进展 2012年7期
牛怀成,李利春,李 瑛,韩文峰,唐浩东,刘化章
(浙江工业大学工业催化研究所,浙江 杭州310032)
进展与述评
高比表面积氟化镁的合成及其在催化中的应用研究进展
牛怀成,李利春,李 瑛,韩文峰,唐浩东,刘化章
(浙江工业大学工业催化研究所,浙江 杭州310032)
综述了氟化镁的合成、表征及在催化中的应用,指出了氟化镁在催化应用中的优缺点。氟化镁具有耐腐蚀性、高稳定性、表面性质可调等优点。目前主要应用的催化体系有:氟氯交换反应、氯氟烃的歧化反应、氯氟烃的加氢脱氯反应、加氢反应、加氢脱硫反应、氮氧化物的脱除等,在具有腐蚀性气体及反应介质的催化反应体系中具有独特的优势。缺点是氟化镁比表面积较小和表面酸性较弱,限制了其在催化研究中的应用。总结了高比表面积氟化镁及负载固体酸催化剂的制备方法及其在各类反应中的应用现状,重点阐述了高比表面积氟化镁的孔结构及其表面酸性的调控方法,最后指出了氟化镁催化剂在ODS替代物的合成领域及其它方面的发展前景。
氟化镁;固体酸催化剂;高比表面积;氟氯交换反应
氟化镁是白色四方粉晶,微溶于水,不溶于乙醇,可以溶于硝酸溶液,是金红石结构,空间点群为P42/mnm。晶格中的每个氟离子周围有3个镁离子,同时镁离子存在于由6个氟离子构成的带缺陷位的八面体中。由于本身的特殊结构,氟化镁在很多领域具有广泛的应用,可以用于制造陶瓷、玻璃;冶金工业中用作助熔剂;光学中用作涂层、膜材料[1]或荧光材料。同时氟化镁具有耐腐蚀性、高稳定性等特点,可以作为催化剂或载体用在具有特殊环境的催化反应中,如含腐蚀性气体氟化氢的反应。本文对高比表面积氟化镁的合成及作为载体负载固体酸催化剂在催化中的应用进行了总结,对氟化镁作为催化剂或载体的制备方法的特点和优劣进行了对比,并阐述了氟化镁在催化领域应用的发展动态,总结了氟化镁催化剂或载体应用于各类反应中的特点及应用过程中的不足,希望为该领域的研究提供参考。
1 氟化镁晶体的制备方法
氟化镁作为一种无机盐,具有特定的晶体结构。目前,不同的氟化镁生产企业根据配套企业所具有的各种优势不同,如周围富产氟资源等,而采用不同的氟化镁生产工艺[2]。目前市场上销售的氟化镁,一般是用作助熔剂、光学镜片等,对比表面积没有很大的要求。通常采用的生产方法为沉淀法:根据采用镁源的不同,可以分为碳酸镁法、硫酸镁(或硝酸镁)法、氧化镁法和氯化镁法等。最常见的是由氢氟酸和菱镁矿粉[3-4]作为原料的生产工艺。该工艺设备简单、易于操作、生产稳定、能耗低、投资少,且装置三废处理措施完善,基本不造成环境污染,具有较好的经济效益和社会效益。
由氢氟酸和菱镁矿粉制取氟化镁的主要化学反应式如式(1)~式(3)所示。

这种方法生产的 MgF2是近于中性的固体物质,结晶度较高,表面几乎没有酸性位,而且其比表面积小,不能应用在催化领域[5]。氟化镁如果要应用在催化领域,必须具备高的比表面积。当氟化镁的比表面积较高时,氟化镁往往以不规则形态存在从而产生了相当数量的缺陷位。表面上镁离子的配位未饱和产生了相当数量的Lew is酸位,呈现中等强度的路易斯酸性[6],提供了高的比表面积和合适的酸性位的强度和数量。因此提高氟化镁的比表面积对拓展其在催化领域的应用具有重要的意义。
2 高比表面积氟化镁的制备方法
高比表面积氧化物的制备是目前材料领域的一个研究热点,常用的方法有模板法、溶胶凝胶法、微乳液方法等[7]。但是对于氟化物,特别是高比表面积氟化镁体系的研究相对较少,大部分报道来自德国洪堡大学的Kemnitz研究组。表1总结了目前文献报道的高比表面积的氟化镁合成方法。
2.1 溶胶-凝胶氟化法

表1 高比表面积氟化镁的制备方法及性质
溶胶-凝胶法一般是首先将原料分散在溶剂中,经过水解反应生成单体,单体进行聚合,开始成为溶胶,进而生成具有一定空间结构的凝胶,经过干燥和热处理制备出所需的纳米粒子。此法制备的材料一般都具有较高的比表面积、可修剪的形貌及纳米粒子尺寸。无水溶胶-凝胶氟化法可以用于制备高比表面积金属氟化物:金属醇盐与无水氟化氢发生反应后,金属离子与氟离子聚合形成M—F—M键并在合适的条件下形成了由纳米金属氟化物粒子构成的稳定的溶胶,随着时间的延长,这些纳米粒子就会彼此串联在一起形成三维空间结构的凝胶,除去凝胶内部的溶剂,并焙烧后即可得到多孔的高比表面积金属氟化物。无水溶胶-凝胶氟化法制备氟化镁一般选用甲醇镁与无水氟化氢在有机溶剂中反应。当氟化氢的含量低于化学计量比时,氟化反应生成了以 Mg(OCH3)2-xFx为通式的含醇盐的镁氟化物,而且水解后分别生成对应的氢氧氟化物/氧氟化物。完全氟化后生成具有高比表面积的氟化镁。金属氟化物的生成过程如图1所示。
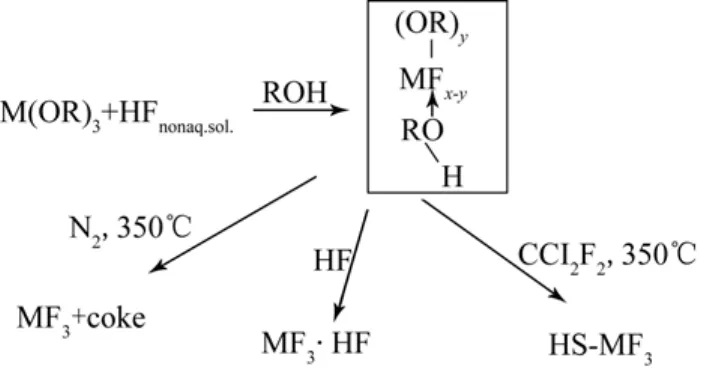
图1 高比表面积金属氟化物及掺杂氟化物的制备过程[8]
通过溶胶-凝胶氟化法制备的氟化镁是无定形的,具有较高的比表面积(150~350 m2/g),普通氟化镁的比表面积只有0.4 m2/g[9]。图2给出了由甲醇镁和无水氟化氢制备得到的高比表面积氟化镁的氮气吸附-脱附等温线及孔径分布图,通过该法合成的氟化镁比表面积可以达到218 m2/g[10]。
Wojciechowska等[11]发现,在MgF2中混合不同比例的 MgO可以进一步提高所制备的样品的比表面积。他们通过一步溶胶-凝胶法制备得到不同比例的MgF2和MgO混合载体,结果显示,当MgO 含量在 71%时,所得到的载体比表面积可接近 450 m2/g,而且能较好保持载体的缺陷位,同时其催化性能也远远好于单纯用MgF2或者MgO负载的催化剂。
文献[6]报道了金属醇盐与氢氟酸水溶液的反应,氟化与水解反应会同时进行,由于氟化反应的速度远远大于水解反应的速度,经过较短时间的反应,氟化反应成为主导反应,生成含氟量较高的产物MgF2-x(OH)x,同时也可以通过调变水与氟化氢的比例,得到不同氟含量的含羟基高比表面积金属氟化物,反应如式(4)所示。

图2 高比表面积氟化镁的氮气吸附-脱附等温线及孔径分布图[9](1 Å=0.1 nm)
Stefan等[12]报道了氟化-水解两步法制备不同羟基含量的MgF2-x(OH)x,在一定温度下焙烧处理,生成高比表面积的金属氧氟化物(图3)。
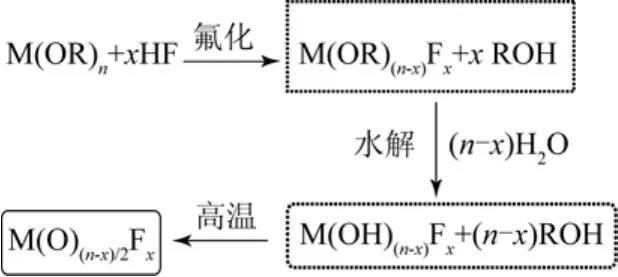
图3 金属氧氟化物的合成图[6]
2.2 硬模板法
硬模板法是近期发展起来的制备多孔材料的新方法。它的基本原理是利用纳米材料作为模板合成材料,然后通过一定的方法把模板除掉,得到多孔材料。材料的孔径大小可以通过模板的纳米粒子大小或者模板的孔结构来调控。利用硬模板法可以得到孔径可调的高比表面积的催化材料,基本原理如图4所示。

图4 硬模板法合成多孔材料的原理示意图[13]
根据硬模板法制备的多孔材料如氧化硅、多孔碳、氧化镁、氧化铁等,并已经成功应用到催化剂及吸附等领域[14-19]。最常用的模板是氧化硅和单质碳。通常氧化硅模板可以采用氢氟酸或者氢氧化钠碱溶液除去,多孔碳模板则可以采用空气气氛下焙烧的方法除掉。
蛋白石是一种含天然水溶性二氧化硅的矿物质,由粒径在150~400 nm的球形二氧化硅粒子规则排列而成。反蛋白石可认为是蛋白石的反向复制体,即将蛋白石中的球形二氧化硅除去后所剩余的充满球形空隙的框架结构。这些规则排列的蜂窝状孔增加了催化材料的比表面积。文献[20-21]报道了以蛋白石为模板合成高比表面积氟化镁。首先要制备类蛋白石模板,其粒子尺寸对应着目标反蛋白石的孔径尺寸,然后将模板中间空隙用氟化镁溶胶填满,待前体固化后除去模板,就可以得到高比表面积的氟化镁。以PMMA为硬模板合成高比表面积氟化镁为例细述外模板法的应用,PMMA悬浊液经离心干燥后作为模板(图 5),用溶胶-凝胶法制备的氟化镁溶胶充分浸渍PMMA模板,干燥后一定温度下焙烧处理就可得到高比表面积氟化镁。
吕剑等[17]报道了采用Mg、Al、Zn、Ni等金属粉末作为铬基氟化催化剂的助剂组分,再与HF反应生成氟化物,产生多孔结构,增加了催化剂的微孔比例,改善了催化剂的机械强度和抗晶化能力,抑制了六价铬生成,避免活性组分铬的流失,催化剂的性能得到大幅度的改善。此法实际上与硬模板法造孔为一个思路,但是用活泼的金属粉末造孔,首先需要采用机械混合的方法将金属粉末掺杂到催化剂前体中,很难做到纳米尺度上的均匀混合。
2.3 氧化分解法
部分含氟有机金属盐的热分解也可以制备二元金属氟化物,但这个反应需要在相当高的温度下才可以实现,高温条件下容易引起材料的烧结,不利于形成高比表面积。含氟金属氟化物在较低温度的热分解利于产生高比表面积氟化物,Skapin等[22]采用了另一种方法,在室温下用对应金属盐与强氧化剂F2反应,氧化分解得到高比表面积材料:室温下,Mg(NO3)2·2H2O与F2在无水液态的HF中发生氧化分解生成了部分结晶的MgF2,比表面积为70 m2/g。反应式如式(5)所示。

图5 PMMA基模板及MgF2的扫描电镜图[21]

2.4 凝胶-模板法
文献[23]中曾报道了采用凝胶-模板法制备高比表面积氧化镁,以四水乙酸镁为前体,以大米粉形成的凝胶为模板,采用新的凝胶-模板法制备了一系列的氧化镁材料,该方法主要利用大米粉在水中加热形成凝胶来分散镁盐前体,再通氧焙烧去除模板从而获得多孔MgO材料。结果表明,制得的MgO具有高比表面积(可达206 m2/g)和双介孔结构(孔径分别位于3.9 nm和5~40 nm附近)。类推高比表面积氟化镁的制备:以四水乙酸镁为前体,以大米粉形成的凝胶为模板,大米粉在水中加热形成凝胶分散镁盐前体,在通氧焙烧除去模板之前用HF气相氟化,有望制备得到高比表面积氟化镁。该方法既不像溶胶-凝胶法需要昂贵的醇盐和大量的有机溶剂,也不像外模板法对模板的孔结构等性能有较高要求,因而有可能发展成为一种制备成本较低且环境友好的多孔材料的新方法。
2.5 微乳法
Saberi等[24]报道了在水与环己烷的微乳液中采用共沉淀法制备得到了粒径在9~11 nm,比表面积为190 m2/g的氟化镁粉末,但由于不能除尽产物中的残留有机物造成了产物在应用上存在局限性。Okuyama等[25]报道了控制氟化镁粒径的最佳方法,通过调控 NH4F/MgCl2的摩尔比制得了粒径范围在6~300 nm且粒度可调的球状和立方体形貌的粒子。
3 高比表面积氟化镁在催化领域中的应用
Lew is 酸通常作为催化剂应用于氟化反应中[26],提高多相催化剂表面的Lew is 酸性就显得更为重要。尽管MgF2表面存在Lew is 酸位[27],但规则结晶态的 MgF2表面的酸性很弱,即普通 MgF2的表面Lew is 酸位少、酸性弱,不能在催化领域得以广泛应用。高比表面积 MgF2结晶度较低并以不规则形态存在,产生了相当数量的缺陷位,表面上的镁离子配位未饱和,一般以三配位、四配位、五配位形式存在。而材料表面的Lew is酸活性位数及Lew is酸强度取决于镁离子的已配位数,配位数越少,材料表面的 Lew is酸活性位数目越多、Lew is酸性越强。高比表面积 MgF2的特殊配位数使得材料本身具有相当数量的 Lew is酸活性位(5~6 sites/nm2)和中等强度的Lew is 酸性[28],这有利于高比表面积 MgF2在催化领域中的应用。研究人员对不同预处理温度下的HS-MgF2进行了NH3-TPD表征[9](图 6),并发现 HS-MgF2表面具有较强的Lew is酸位,而且在不同的预处理温度下表面的酸量会有较大的差别:预处理温度在 120 ℃时,表面的酸量较多;300℃预处理后,表面酸量明显减少;预处理为500 ℃时,表面酸量几乎为0。可能原因是在500 ℃高温处理后,氟化镁高度结晶,表面酸量明显减少。
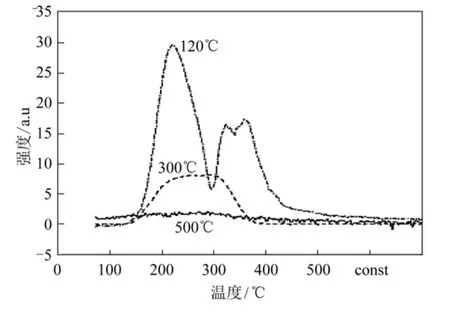
图6 不同预处理温度下HS-MgF2的NH3-TPD曲线 [9]
表面镁离子的配位未饱和使得高比表面积氟化镁呈现了中等强度的Lew is酸性,但用于催化一些有机反应时表面酸性可能还太弱。为增强高比表面积氟化镁的酸性,拓宽它在固体酸催化反应中的应用,通常采用引入其它三价金属离子的方式来调变氟化镁的酸性。
3.1 氟化镁负载催化剂的制备方法
以铬镁催化剂体系Cr-MgF2的合成为例,概述以MgF2为主体相的M-MgF2掺杂体系的合成方法。有研究发现,气相氟化反应中Cr-MgF2催化剂有着与 Cr-A lF3相似的催化性能,而且催化剂的寿命较长,在气相氟氯交换反应中Cr-MgF2催化剂具有更广阔的应用前景。
3.1.1 溶胶-凝胶法
金属离子的掺杂会改变固态金属化合物的特性。对于金属氧化物,这种特性的改变已经通过Tanabe’s 模型[29]得以解释。这种模式已经推广到了金属氟化物中[26]。根据 Tanabe’s 模型氟化物中的Mn+离子被 M(n+1)+部分取代,在 M(n+1)+位上多出了一个正电荷产生了Lew is酸位。必须先保证掺杂离子与被掺杂离子的尺寸相近及合适的离子浓度,掺杂的摩尔分数在不高于30%时才不会改变主体晶格结构并能做到完全相容[29]。无水溶胶-凝胶氟化法就提供了一种方便的金属离子掺杂氟化镁的制备方法,采用这种混合方法可以做到掺杂的金属组分能够均匀地分散到高比表面积的主体金属氟化物中。
在M-MgF2掺杂体系中,掺杂的金属化合物一般都是金属醇盐,如果金属醇盐不容易得到,也可以使用其它的前体。HS-MgF2中镁离子尺寸(0.72 Å)适中,适合很多三价金属离子的掺杂,掺杂在M-MgF2体系中的三价金属离子可以是Cr[30]、Ga[8,10]、In[8]、Fe[31]、V[26](图9),溶胶-凝胶氟化制备的掺杂氟化镁催化剂的比表面积要比沉淀法制备的材料比表面积高。
吡啶吸附红外光谱法可以表征催化剂表面的不同酸位,文献[26]的研究指出,M-MgF2掺杂体系的Lew is酸位不是来自于氟化镁,而是来自于掺杂进的金属离子,即掺杂的三价金属氟化物增加了催化剂体系表面的Lew is酸位(图7是掺杂不同含量VF3后催化剂的吡啶吸附的红外光谱图)。
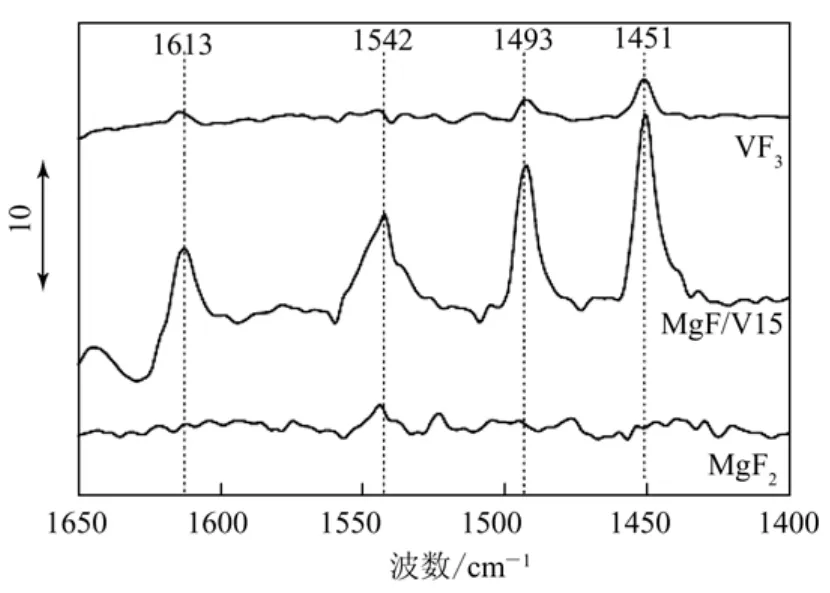
图7 VF3/MgF2上吡啶吸附的傅里叶变换红外光谱图[26]
文献[30]报道了铬掺杂的氟化镁催化剂,研究了不同铬的前体及铬的掺杂量对溶胶-凝胶方法合成的铬镁催化剂的影响,对样品的酸性及比表面积进行了表征,并评价了该系列催化剂在CF3-CHClF的歧化反应中的催化性能。研究发现,向二价镁氟化物中掺杂三价金属离子可以提高其Lew is酸性,歧化反应性能和催化剂的 Lew is 酸性能相关。CF3-CHClF的歧化反应如式(6)所示。

3.1.2 沉淀法
沉淀法尤其是共沉淀法在催化剂的制备中应用比较广泛,载体和活性组分在沉淀剂的作用下得到共沉淀产物,经焙烧处理得到所需催化剂。文献[32]报道了 Cr/MgF2的制备方法及催化应用。镁与铬的硝酸盐溶于一定量乙醇中,将该溶液逐滴加入到40%氢氟酸溶液中反应3 h,离心、洗涤、干燥、焙烧,得到不同铬镁比例的催化剂。也可以通过在MgF2中加入可溶性铬盐溶液形成悬浊液,于搅拌下加入沉淀剂(一般用NH3·H2O)生成Cr(OH)3沉淀,经过滤、干燥、成型制得催化剂前体,最后活化处理制成Cr-MgF2催化剂。Cho等[33]制备了一系列的Cr-MgF2负载型催化剂,其中MgF2由氧化镁和过量的氢氟酸制备。
沉淀法制备的各组分分布均匀,催化剂性能稳定,主要特点是活性组分和载体的结合力比浸渍法制备的材料结合力要强,反应中活性组分流失少。在制备过程中,沉淀剂的选择、溶液的浓度、沉淀温度、pH值、搅拌速度和加料顺序都影响沉淀的性质。沉淀条件不同,所得催化剂的孔结构、比表面积、晶形和机械强度均不同。因此,催化剂的重复再现性差,实验操作要求较高。
3.1.3 浸渍法
浸渍法制备M-MgF2催化剂,是用Cr或其它组分的可溶盐溶液通过浸渍法引入到 MgF2表面。通过控制溶液的浓度、温度和浸渍时间来调节活性组分在载体表面的分布。活性组分以离子或化合物的形态负载于载体表面,经无水HF活化制得氟化催化剂。由于活性组分之间的竞争吸附和浸渍液溶质的迁移,催化剂的活性组分分布不均匀,稳定性差。而且浸渍量较少,易流失,难以达到预定的负载量,催化活性低[34]。
3.1.4 共混法
根据混合时原料的状态,共混法可分为干混和湿混。干混法是将Cr2O3和载体按一定比例混合、研磨、捏合成型,通过焙烧、活化制成氟化催化剂。湿混法是将载体加入活性组分水溶液中,在连续搅拌下滴加NH3·H2O,使活性组分沉积在载体表面,制成相应的催化剂前体。吕剑等[35]报道了以碱式碳酸镁和氢氟酸制备MgF2,以CrCl3·6H2O 和氨水制备 Cr(OH)3·H2O,将二者混合均匀即制得催化剂前体,用HF活化后即得Cr-MgF2催化剂[36]。专利US5672789报道[37]将Cr(OH)3·H2O和MgCl2·6H2O的水溶液混合后,加入氢氟酸反应,经过滤、干燥和焙烧,制得 Cr-MgF2催化剂前体。专利 GB 229555[38]中提出可将 CrO3、MgF2、CaF2、CeF2、ZnF2和 H2O在甲醇或乙醇中回流,制成氟化催化剂。文献[39]也报道了以Cr(NO3)3·9H2O、MgO、石墨和水混合也可制得Cr-Mg催化剂。共混法的优点是操作简单,已成为工业制备氟氯交换催化剂的首选方法。
3.2 氟化镁本体及负载催化剂的应用
HS-MgF2表面的中强酸可以催化部分有机反应[10,40],如CHClF2的歧化反应[(式(7)],转化率在350 ℃时达到60%。
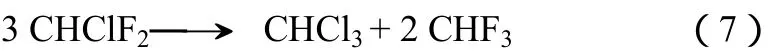
文献[40]报道了氟化镁作为催化剂可以高效高选择性催化3-氯-1,1,1,3-四氟丁烷的脱卤化氢反应,结果见图 8。氟化镁可以选择性地催化含卤化合物的脱氟化氢反应,并抑制了脱氯化氢反应的进行。3-氯-1,1,1,3-四氟丁烷在氟化镁表面反应120 min后,反应物的转化率为83%;反应速率为0.30 µmol/s·m2;脱氟化氢反应的选择性为90%;脱氯化氢反应的选择性为10%。

图8 不同催化剂上3-氯-1,1,1,3-四氟丁烷的转化率随时间的变化关系[40]
尽管HS-MgF2可以催化某些有机反应,但文献[27]指出HS-MgF2更适合于作为载体负载催化剂,通常氟化镁作为载体负载铬、铁、镍等催化活性组分,载体氟化镁和活性组分的相互作用提高了催化剂的活性。应用的反应体系主要有以下几种。
(1)氟氯交换反应 气相催化氟氯交换反应是合成全氟或者氢氟代烃的重要过程。反应在 HF气流中进行,金属氧化物催化剂在反应条件下完全氟化,转变为氟化物催化剂。因此氟化物催化剂在氟氯交换反应中显示了其唯一可在酸性介质中使用、再生的特性,在合成对大气臭氧层无破坏替代物方面,具有其它类催化剂无法取代的地位。
氟氯交换反应包括CFCs的氢氟化、歧化等反应,见式(8)~式(12)。
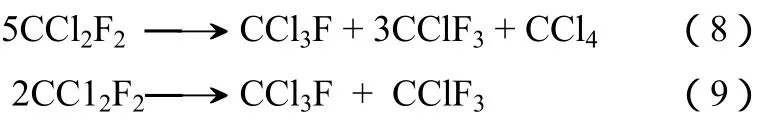

气相氟化反应广泛采用铬基催化剂,主要活性组分为铬的氟化物或氟氧化物;主要载体为活性炭、氧化铝和氟化铝等。目前 Cr-AlF3催化剂的活性较高,故Cr-A lF3催化剂的应用最为广泛。但Cr-A lF3的强酸性导致催化剂易失活。Lee等[41-42]报道了镁基载体(MgF2或MgO)负载活性组分在气相合成氢氟烃反应中的应用,认为 MgF2是较好的催化剂载体。尽管 MgF2本身活性不高,甚至在某些催化反应中没有催化活性,但和活性组分Cr混合后大大改善了催化剂的催化性能,即高比表面积氟化镁负载活性组分后增大了催化剂的比表面积、分散度、Lew is酸性,提高了催化剂的反应活性。
以R12的歧化反应[(式9)]为例简述氟化镁负载催化剂对氟氯交换反应的催化作用。Kemnitz等[32]对不同镁含量Cr-MgF2催化剂的活性进行了评价(图9),他们发现:当镁的摩尔分数在56%以上时,CC12F2的转化率在60%以上(纯的MgF2除外,因为100%的MgF2本身酸性太弱不足以催化该歧化反应),是CC12F2歧化反应的高效催化剂。调控不同的铬镁摩尔比例即可调变催化剂表面的L酸位,改善催化剂的催化性能。
载体氟化镁与活性组分的相互作用,提高了催化剂的活性和选择性,延长了催化剂的使用寿命[27]。活性组分可以通过溶胶-凝胶法、沉淀法、浸渍法、共混法负载到氟化镁载体上。
Kemnitz等[10,31]用不同Fe3+前体通过两步无水溶胶-凝胶法合成了Fe-MgF2催化剂,研究了FeCl3、 Fe2(SO4)3、Fe(OCH3)3三种前体的影响,并对催化剂的酸性及比表面积进行了表征,同时报道了该催化剂在CCl2F2的歧化反应及C2Cl4氢氟化反应中的催化性能。研究指出:以Fe(OCH3)3作为Fe3+前体,用HF进行低温氟化得到的催化剂表面的Lew is酸性要低于用CCl2F2氟化的催化剂,后者在C2Cl4的氢氟化反应及 CHF2CHF2的异构化反应中活性不高[10]。MgF2负载的 Fe3+催化剂 Fe-MgF2对反应式(10)有高效催化作用,而且氟化镁负载Fe3+催化剂Fe-MgF2对反应式(12)中反应物的转化率达到100%[10]。同样,可以利用制备Cr-MgF2的方法,如溶胶-凝胶法、沉淀法、浸渍法等制备其它的M-MgF2催化剂,并对其应用进行研究。

图9 不同镁含量的Cr-MgF2催化剂对CCl2F2转化的影响

图10 高表面积氟化镁及负载三价金属氟化物的催化剂的NH3-TPD图[43]

表2 反应温度为623 K时,在不同催化剂上CHClF2歧化反应的转化率
文献[43]报道采用高比表面积氟化镁负载氟化铝、氟化铁制备了一系列催化剂,三价金属离子的掺杂明显提高了高比表面积氟化镁的 Lew is酸性(图 10),更有利于固体酸催化反应的进行。同时研究了掺杂前后的催化剂上 CHClF2歧化反应的活性。表2给出了催化剂对CHClF2歧化反应的活性数据。可以看出无掺杂时,HS-MgF2对CHClF2的转化率只有41%;掺杂后催化剂对反应物的转化率都有所提高,尤其是掺杂铁离子后转化率达到了100%。
MgF2负载不同三价金属离子(V3+、Fe3+、Ga3+、In3+)的催化剂[8]也可以催化CC12F2的歧化反应(图11),且Ga3+摩尔负载量在20%时,反应活性最高。In3+的负载量在 5%时,催化活性最高。VF3/MgF2催化剂对CCl2F2的歧化反应也有较好的活性[10]。

图11 MgF2负载Ga3+及In3+催化剂在CCl2F2歧化反应中的催化活性[8]
(2)加氢反应 近期有文献报道氟化镁可以作为载体负载镍催化甲苯[44]和苯[45]的加氢反应,催化活性也较好(图12)。从图12可以看出,焙烧后的氟化镁负载镍催化剂活性要远远高于氟化铝负载的镍催化剂(Ni/Al-S-R)的反应活性。
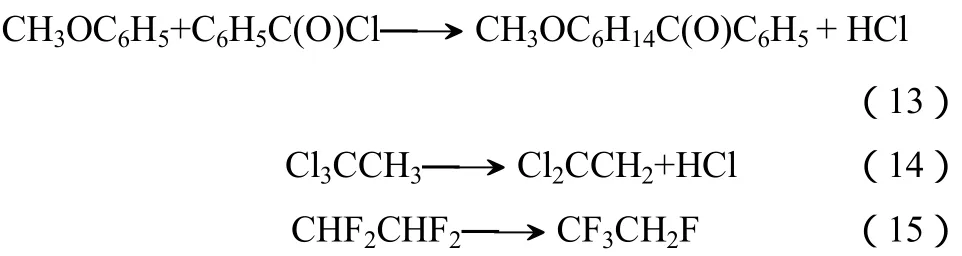
(3)在其它类型反应中的应用 氟化镁负载催化剂还可以催化其它类型的反应,如以一氧化碳和碳氢化合物作为还原剂还原氮氧化物[27];氟化镁作为载体负载钌催化剂后在加氢脱硫反应中表现出了高活性[46]。氟化镁负载的钌或钯催化剂可高效催化氯氟烃的加氢脱氯反应[47-48]。部分催化反应式见式(13)~式(15)。

图12 不同载体负载的镍催化剂对甲苯加氢反应活性的影响[44]
氟化镁负载的催化剂对以上脱氯及异构化反应都有较好的催化作用,其中有些反应的转化率达到 100%[10],这些报道进一步表明了氟化镁作为催化剂的载体具有潜在的应用前景。
4 结 语
氟化镁应用在催化领域,自首次报道以来就受到了广大研究者的关注,尤其是溶胶-凝胶氟化法和模板法的出现,有效提高了氟化镁的比表面积,展现了氟化镁在催化领域的应用前景。科研人员一直致力于高比表面积氟化镁负载固体酸催化剂的制备方法研究、表面酸性调控及其在化工催化等领域的应用。近年来,对固体酸催化剂在 ODS替代物的合成领域的应用得到了较大的进展,而且已经实现了工业化应用。相信在今后的几年内仍然是人们的研究热点之一。
[1] 沈静飞,王海波,黄如喜,等. BaMgAl10O17:Eu2+蓝色荧光粉表面包覆 MgF2的性能[J]. 化工进展,2007,26(12):1771-1775
[2] 徐金尧,明大增,李志祥,等. 氟化镁的制备方法[J]. 无机盐工业,2011,43(9):8-10.
[3] 胡庆福,李保林,李国庭,等. 氟化镁新产品的开发[J]. 河北轻化工学院学报,1993,14(1):27-31.
[4] Bonneau L. Process for preparation of magnesite,application in preparation of magnesium fluoride,and magnesium fluoride thereform:CN,1046511 [P]. 1990-10-31.
[5] 胥会祥,吕剑. 镁基气相氟化催化剂的研究进展[J]. 工业催化,2002,10(6):43-47.
[6] Wuttke S,Coman S M,Krohnert J,et al. Sol-gel prepared nanoscopic metal f uorides——A new class of tunable acid–base catalysts [J].Catal.Today,2010,152:2-10.
[7] Svensson E E,Nassos S,Boutonnet M,et al. M icroemulsion synthesis of MgO-supported LaMnO3for catalytic combustion of methane [J].Catal.Today,2006,117:484-490.
[8] Murthy J K,Groβ U,Rudiger S,et al. M ixed metal f uorides as doped Lew is acidic catalyst systems:A comparative study involving novel high surface area metal f uorides [J].J.Fluor.Chem.,2004,125:937-949.
[9] Murthy J K,Groß U,Rudiger S,et al. Sol-gel-f uorination synthesis of amorphous magnesium f uoride [J].J.Sol.State.Chem.,2006,179:739-746.
[10] Rudiger S,Groß U,Kemnitz E. Non-aqueous sol-gel synthesis of nano-structured metal f uorides [J].J.Fluor.Chem.,2007,128:353-368.
[11] Wojciechowska M,Wajnert A,Tomska-Foralewska I,et al. Properties of magnesium oxo-fluoride supports for metal catalysts [J].Catal.Lett.,2009,128:77-82.
[12] Wuttke S,Coman S M,Scholz G,et al. Novel sol-gel synthesis of acidic MgF2-x(OH)xmaterials [J].Chem.AEur.J.,2008,14:11488-11499.
[13] Lee J,Kim J,Hyeon T. Recent progress in the synthesis of porous carbon materials [J].Adv.Mater.,2006,18:2073-2094.
[14] Liang C D,Li Z J,Dai S. Mesoporous carbon materials:Synthesis and modification [J].Angew.Chem.Int.Ed.,2008,47:3696-3717.
[15] Lei Z,Xiao Y,Dang L,et al. Graphitized carbon w ith hierarchical mesoporous structure templated from colloidal silica particles[J].Micropor.Mesopor.Mater.,2008,109:109-117.
[16] Stein A,Wan Z,Fierke M A. Functionalization of porous carbon materials w ith designed pore architecture [J].Adv.Mater.,2009,21:265-293.
[17] 吕剑,张伟,王磊,等. 氟化催化剂及其制备方法和用途:中国,1651137 [P]. 2005-08-10.
[18] Zhao X S,Su F,Yan Q,et al. Templating methods for preparation of porous structures[J].J.Mater.Chem.,2006,16:637-648.
[19] Li Y,Feng Z,Lian Y,et al. Direct synthesis of highly ordered Fe-SBA-15 mesoporous materials under weak acidic conditions[J].Micropor.Mesopor.Mater.,2005,84:41-49.
[20] Rudiger S,Kemnitz E. The f uorolytic sol–gel route to metal f uorides—A versatile process opening a variety of application f elds [J].Dalton Trans.,2008(9):1117-1127.
[21] Schuth F. Endo- and exotemplating to create high-surface-area inorganic materials [J].Angew.Chem.,Int.Ed.,2003,42:3604-3622.
[22] Skapin T,Tavcar G,Bencan A,et al. Recent developments in the preparation of high surface area metal f uorides [J].J.Fluor.Chem.,2009,130:1086-1092 .
[23] 马丽,蒋平,孙瑞琴,等. 凝胶-模板法制备高比表面积氧化镁[J].催化学报,2009,30(7):631-636.
[24] Saberi A,Negahdari Z,Bouazza S,et al. Synthesis and characterization of crystalline nanosized MgF2powderviam icroemulsion route [J].J.Fluor.Chem.,2010,131:1353-1355.
[25] Nandiyanto A B D,Iskandar F,Ogi T,et al. Nanometer to submicrometer magnesium fluoride particles w ith controllable morphology[J].Langmuir,2010,26 (14) :12260-12266.
[26] Kemnitz E,Zhu Y,Adamczyk B. Enhanced Lew is acidity by aliovalent cation doping in metal f uorides [J].J.Fluor.Chem.,2002,114:163-170.
[27] Wojciechowska M,Zielinsky M,Piotrowski M. MgF2as a non-conventional catalyst support[J].J.Fluor.Chem.,2003,120:1-11.
[28] Kemnitz E,Wuttke S,Coman S M. Tailor-made MgF2-based catalysts by sol-gel synthesis [J].Eur.J.Inorg.Chem.,2011,2011(31):4773-4794.
[29] Tanabe K,Sum iyoshi T,Shibata K,et al. A new hypothesis regarding the surface acidity of binary metal oxides [J].Bull.Chem.Soc.Jpn., 1974,47:1064-1066.
[30] Murthy J K,Groß U,Rudiger S,et al. Synthesis and characterization of chrom ium(III)-doped magnesium f uoride catalysts[J].Appl.Catal.A,2005,282:85-91.
[31] Murthy J K,Groß U,Rudiger St,et al. FeF3/MgF2:Novel Lew is acidic catalyst systems [J].Appl.Catal.A,2004,278:133-138.
[32] Adamczyk B,Hess A,Kemnitz E. Magnesium- and iron-doped chromium fluoride/ hydroxyfluoride:Synthesis,characterization and catalytic activity [J].J.Mater.Chem.,1996,6(10):1731-1735.
[33] Cho D H,Yim S D,Cha G H,et al. Behavior of chromium oxide on MgO or MgF2[J].J.Phys.Chem.A,1998,102:7913-7918.
[34] 张学良. 气相氟化法合成二氟甲烷的研究进展 [J]. 化工生产与技术,2004,11(6):7-10.
[35] 吕剑,胥会祥. Cr3+/MgF2氟化催化剂的制备及其对合成二氟甲烷反应的催化性能 [J]. 催化学报,2003,24(5):379-384.
[36] 胥会祥,吕剑. Cr/ MgF2氟化催化剂活性物种的研究 [J]. 催化学报,2002,23(4):345-348.
[37] Kim H S,Lee B G,Kim H,et al. Catalyst for fluorination of 1,1-dichloro-1-fluoroethane and process for the preparation of 1,1,1-trifluoroethane using the same: US,5672789 [P]. 1997-09-30.
[38] Lee H,Jeong H D,Chung Y S ,et al. Catalyst for fluorination of 1,1,1-trifluoro-2,2-dichloroethane and method for preparing the same:GB,2295556 [P]. 1996.
[39] Wanzke W,Siegemund G,Schm ieder W. Method to the preparation of 1,1,1,2-Tetrafluorethan:EP,417680 [P]. 1991.
[40] Teinz K,Wuttke S,Börno F,et al. Highly selective metal f uoride catalysts for the dehydrohalogenation of 3-chloro-1, 1, 1, 3-tetra f uorobutane[J].J.Catal.,2011,282:175-182.
[41] Lee H,Jeong H D,Chung Y S,et al. Fluorination of CF3CH2Cl over Cr-Mg fluoride catalyst:The effect of temperature on the catalyst deactivation[J].J.Catal.,1997,169:307-316.
[42] Lee H, Kim H S, Kim H, et al. Preparation of 1,1,1,2-tetrafluoroethane by catalytic fluorination of 1,1,1-trifluoro-2-chloroethane over CrF3/MgF2-A lF3[J].J.Mol.Catal.A:Chem.,1998,136:85-89.
[43] Nickkho-Am iry M,Wuttke S,Kemnitz E. A comparative study of surface acidity in the amorphous,high surface area solids,alum inium f uoride,magnesium f uoride and magnesium f uoride containing iron(Ⅲ) or alum inium(Ⅲ) f uorides[J].J.Fluor.Chem.,2008,129:366-375.
[44] Zielinski M,Wojciechowska M. Studies of new magnesium f uoride supported nickel catalysts for toluene hydrogenation[J].Catal.Today,2011,169:175-180.
[45] Zieliński M,Pietrowski M,Wojciechowska M. The effect of preparation of Ni/MgF2catalysts on the hydrogenation of benzene activity[J].PolishJ.of Environ.Stud.,2009,18(5):965-969.
[46] Wojciechowska M,Pietrowski M,Lomnicki S. Novel supported catalyst for hydrodesulfurization reaction[J].J.Chem.Soc.,Chem.Commun.,1999(5):463-464.
[47] Cao Y C,Jiang X Z,Song W H,et al. Hydrodechlorination of CFC-12 over novel supported palladium catalysts[J].Catal.Lett.,2001,76:1-2.
[48] Malinowski A,Juszczyk W,Pielaszek J,et al. Magnesium fluoride as a catalytic support in hydrodechlorination of CCl2F2(CFC-12)[J].J.Chem.Soc.,Chem.Commun.,1999(8):685-686.
Progress of preparation and catalytic app lication of magnesium fluoride w ith high surface area
NIU Huaicheng,LI Lichun,LI Ying,HAN Wenfeng,TANG Haodong,LIU Huazhang
(Institute of Industrial Catalysis,Zhejiang University of Technology,Hangzhou 310032,Zhejiang,China)
This review outlines the synthesis,characterization and catalytic application of MgF2(agnesium fluoride) based materials. The advantage and disadvantage are summarized. Such materials can be used in the systems involving corrosive gases and solvent due to its corrosion resistance and high stability. The catalytic reactions,are chlorine/fluorine exchange reactions,hydrogenation,hydrodechlorination of chlorofluorocarbon,hydrodesulfurization of organic compounds and removal of NOx. The catalytic application of magnesium fluoride itself is lim ited due to its low surface area and weak acidic properties. The recent development on the synthesis,catalytic application of MgF2and supported solid acid catalysts is reviewed. Special attention is paid to the controlled synthesis of MgF2(magnesium fluoride) w ith high specific surface area and tunable acid-base properties. Finally,the importance of research on the preparation of high surface area MgF2and its application prospects in catalysis is pointed out.
magnesium fluoride; solid acid catalysts; high specific surface area; fluorine and chlorine exchange reaction
TQ 426.94
A
1000–6613(2012)07–1484–09
20121-01-10;修改稿日期:2012-03-13。
浙江省钱江人才计划(2010R10039)及国家自然科学基金(20803064)项目。
牛怀成(1986—),男,硕士研究生。联系人:李瑛,研究员,从事多孔材料负载金属催化剂研究。E-mail liying@zjut.edu.cn。
猜你喜欢
安徽农业科学(2023年21期)2023-11-17 02:25:34
云南化工(2021年9期)2021-12-21 07:43:44
陶瓷学报(2020年3期)2020-10-27 02:08:12
饮食保健(2019年15期)2019-08-13 01:33:22
中国卫生标准管理(2015年17期)2016-01-20 09:26:44
中国当代医药(2015年9期)2015-03-01 02:02:03
中国塑料(2014年12期)2014-10-17 02:49:41
应用化工(2014年11期)2014-08-16 15:59:13
应用技术学报(2014年2期)2014-02-28 14:52:22
无机盐工业(2013年1期)2013-03-19 23:25:40
