
糖类化合物的端基效应
2012-09-25 03:39吕萍王彦广
大学化学 2012年2期
吕萍 王彦广
(浙江大学化学系 浙江杭州 310027)
端基效应也叫异头碳效应(anomeric effect),是糖化学中常见的一种作用,而且是一类非常有趣和重要的立体化学现象。然而,由于端基效应与取代环己烷的优势构象分析的结果相矛盾,从而导致这方面的知识成为基础有机化学教学中的一个难点。为此,本文就端基效应的产生、理论解释及相关应用等进行简要介绍,以期为相关教学和学生自学提供帮助。
1 问题的提出
许多有机化学教材在描述单取代环己烷稳定构象时[1-3],认为取代基处于平伏键(e键)的构象1是优势构象,而构象2由于存在两个1,3-直立键(a键)位阻(1,3-直立键的排斥力)为非优势构象(图1)。
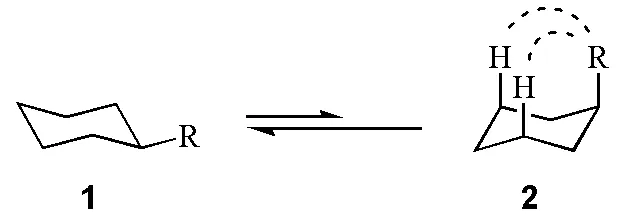
图1 单取代环己烷的两种构象
1,3-直立键位阻的大小取决于基团的种类,如表1所示。根据ΔE=-RTlnK,其中ΔE是两种构象(1和2)的能量差值,R是气体常数(8.315J/(K·mol)),T是绝对温度,我们可以求出平衡状态时两种构象(1和2)的百分比K,结果列于表1。可以看出,取代基的位阻越大,优势构象(1)所占的份额也越大。当取代基为叔丁基时,99.99%的1-叔丁基环己烷以优势构象(1)的形式存在,也就是说叔丁基处于直立键的概率仅为0.01%。

表1 1,3-直立键位阻及构象1和2在平衡状态时的百分比
对于与环己烷结构相似的四氢吡喃环,这一规则仍然适合,即2-烷基四氢吡喃的优势构象是3,而不是4(图2)。
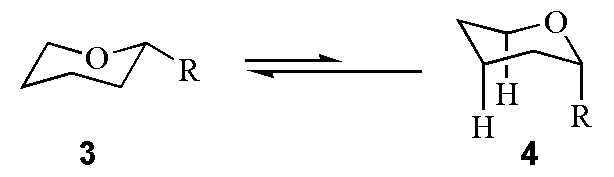
图2 2-烷基四氢吡喃的两种构象
对于葡萄糖而言,通过D-葡萄糖开链形式,β-D-吡喃葡萄糖和α-D-吡喃葡萄糖达到平衡,β-D-吡喃葡萄糖比较稳定,占62.6%;α-D-吡喃葡萄糖则较不稳定,占37.3%;开链形式仅占0.002%(图3)。β-D-吡喃葡萄糖和α-D-吡喃葡萄糖互为非对映异构体。由于不同的手性来自于1号碳,故在糖化学中称为端基异构体。
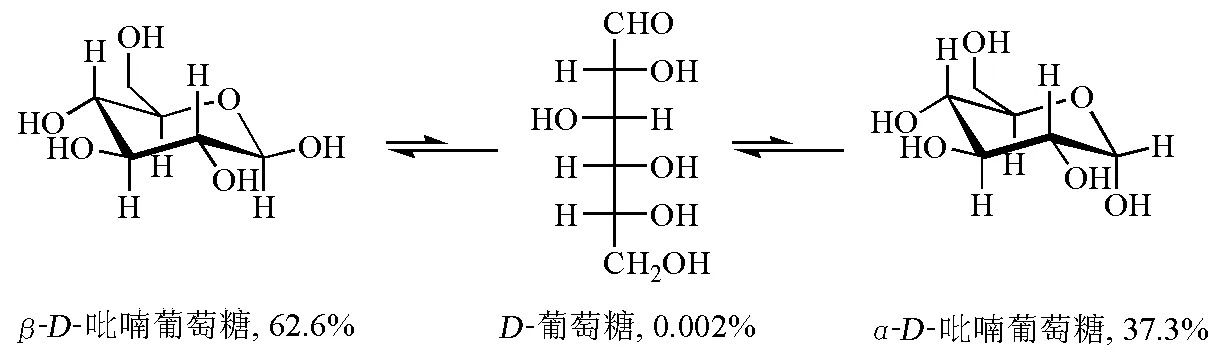
图3 α-D-吡喃葡萄糖和β-D-吡喃葡萄糖构象
D-甘露糖是D-葡萄糖的C-2差向异构体,当它形成吡喃环时,人们发现α-D-吡喃甘露糖比β-D-吡喃甘露糖稳定,尽管α-D-吡喃甘露糖中有两个羟基占据直立键,而β-D-吡喃甘露糖只有一个,但α-D-吡喃甘露糖还是占68%,为优势构象(图4)[5]。这一现象称为糖的端基效应。
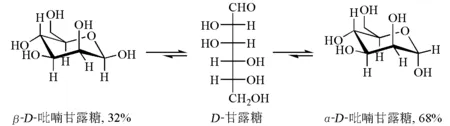
图4 α-D-吡喃甘露糖和β-D-吡喃甘露糖构象
并非只有D-吡喃甘露糖具有端基效应。当人们用醇和质子酸处理β-D-吡喃葡萄糖时,得到两种吡喃葡萄糖苷,即α-D-甲基吡喃葡萄糖苷和β-D-甲基吡喃葡萄糖苷(图5)。有意思的是,α-D-甲基吡喃葡萄糖苷占66%,而β-D-甲基吡喃葡萄糖苷占33%,用醇和质子酸处理α-D-吡喃葡萄糖时得到相同结果。

图5 D-甲基吡喃葡萄糖苷的形成
对于2-杂原子取代的D-吡喃葡萄糖,当端基上的取代基电负性增大时,端基效应更加明显(图6)。端基上的取代基由乙酰氧基改变为氯时,取代基占直立键构象的比例由86%增大到94%。
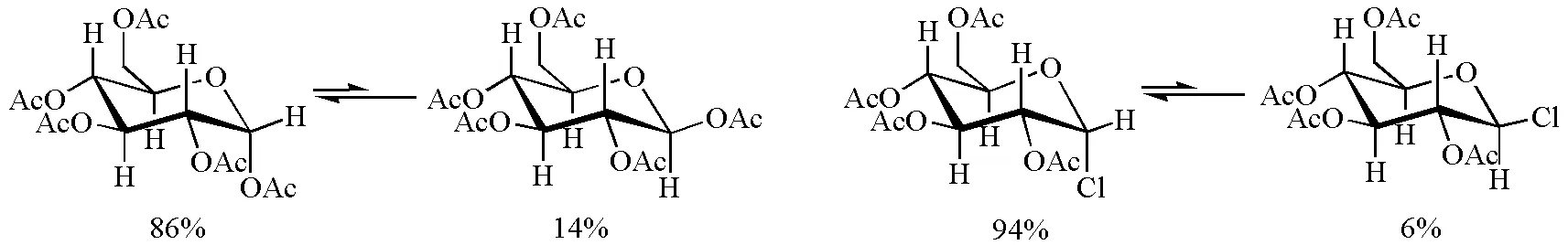
图6 2-杂原子取代吡喃葡萄糖构象组成
糖的端基效应可以衍生到其他2-杂原子取代的六元环体系(图7),杂原子(X,Y)可以是氮、氧、硫、氟、氯、溴、碘。在两种可能的构象(5和6)中,构象5为优势构象。最常见的杂原子有硫、氧和氟。骨架如图7中的7、8、9。

图7 2-杂原子取代杂六元环的两种构象
在一些开链体系中也发现了端基效应。含有Lp—X—C—Y链的化合物(Lp是X原子所带的孤电子对)就属于这种体系。例如,二甲氧基甲烷有两种可能的构象,邻位交叉式和全交叉式,以邻位交叉式为主(图8)。

图8 二甲氧基甲烷的两种构象
更有趣的是,端基效应不仅与分子结构有关,而且与溶剂的极性、温度、浓度等因素有关。通常,溶剂极性越小,端基效应越强。与水相比,乙醇更能体现糖的端基效应。温度越低,端基效应越强。如从冷水中结晶D-葡萄糖得到的是α-D-吡喃葡萄糖;而从热水中结晶D-葡萄糖则得到β-D-吡喃葡萄糖。
为了探索端基效应与溶剂极性的关系,Lemieux等人[6]合成了4,4,5,5-四氘代-2-甲氧基四氢吡喃(图9),研究了该化合物在不同氘代溶剂中的氢谱,从而测定出甲氧基占直立键时构象(10)和甲氧基占平伏键时构象(11)的百分比(表2)。在四氯化碳中,该化合物的端基效应非常明显(构象10占83%);而在水溶液中,两种构象所占的比例差不多。

图9 4,4,5,5-四氘代-2-甲氧基四氢吡喃的两种构象

表2 两种构象在不同氘代溶剂中的百分比
2 端基效应的解释
从2-杂原子取代杂六元环(图7)的Newman投影式(图10)可以看出,构象5中存在两个由邻位交叉引起的排斥力,而构象6只有一个,由此知构象5存在较大的立体位阻。那么构象5的相对稳定性应该如何解释?是否构象5中还存在其他的稳定化能?抑或是构象6中存在其他的排斥力(即去稳定化能)?

图10 构象5和构象6的Newman投影式
2.1 静电排斥力
最直接的解释是静电排斥力。如图11所示,虽然构象5中存在两个邻位交叉所引起的立体位阻,但构象6中存在两个由孤对电子邻位交叉引起的静电排斥[7],这种静电排斥和立体位阻共存于一个分子中,决定着分子的优势构象。
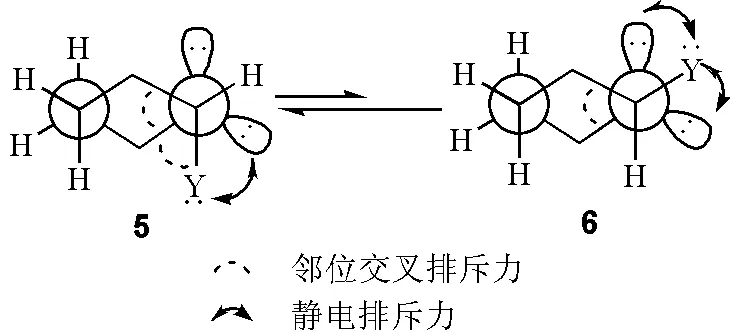
图11 构象5和构象6中的立体位阻和静电排斥力竞争
2.2 偶极矩
端基效应可以用分子偶极矩进行解释。图12显示了构象12和构象13中的3个C—O单键的偶极矩方向。在构象12中,偶极矩为相反的方向,电子云密度被整个体系分散并稳定;与构象13相比,构象12具有更小的分子偶极矩,在非极性溶剂中相对比较稳定,即:溶剂极性越小,端基效应越强。

图12 构象12和构象13的偶极矩
2.3 分子轨道理论
在顺-2,3-二氯-1,4-二氧六环(图13)中,连有直立键氯的C—O键长为1.39Å,连有平伏键氯的C—O键长为1.43Å[8],显然,连有直立键氯的C—O键具有部分双键的性质,而连有平伏键氯的C—O键是经典的C—O单键,前者比后者更稳定。连有氟的C—O也有这样的特性,从图13可以看出,连有直立键氟的C—O键键长(1.382Å)比连有平伏键氟的C—O键键长(1.406Å)短,而且处于直立键的C—F键键长(1.397Å)比处于平伏键的C—F键键长(1.367Å)长[9]。

图13 连有直立键和平伏键杂原子的C—O键长比较
与连有平伏键氯的C—O相比,连有直立键氯的C—O键具有一定的稳定性,这是由于氧原子上的孤对电子可以反充到C—Cl的反键轨道,这种σ*-p超共轭效应使C—O键具有部分双键的性质(图14),同时,C—X键的键长增长,这一假说称为反式共平面孤对电子假说(antiperiplanar lone pair hypothesis,ALPH)。
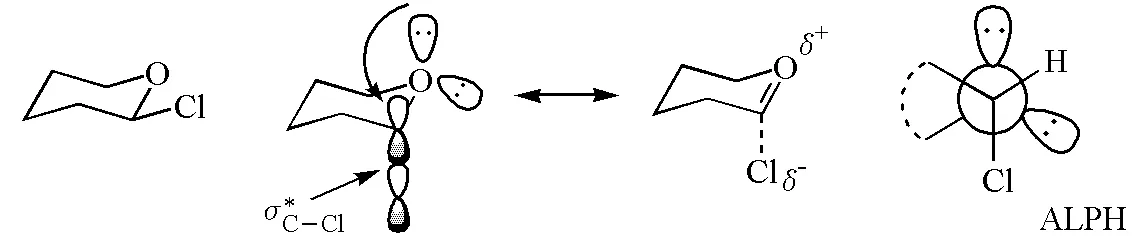
图14 ALPH假说示意图


图15 氘代三丁基锡和2-卤代-四乙酰基-D-吡喃葡萄糖的反应
3 端基效应的应用
3.1 关于有机反应立体选择性的合理解释
1,9-二羟基-壬酮在酸催化下脱水成缩酮(图16)[11-12],该缩酮可以有4种立体异构体(14~17)。其中异构体14有两个端基效应,异构体15和异构体17各有一个端基效应,异构体16没有端基效应,产物以14为主。

图16 端基效应对螺缩酮的选择性控制
芳醛可与二烯发生氧杂Diels-Alder反应,生成的环己烯基硅醚分别在碱性和酸性条件下失去三甲基硅基,得到两种不同的端基异构体产物(图17)[13]。在碱性条件下,甲氧基亲核进攻硅,离去的烯醇负离子发生质子交换生成产物,端基碳原子的手性保持不变。在酸性条件下,当烯基硅醚醇解时,缩醛通过开链转化成更稳定的有端基效应的缩醛。
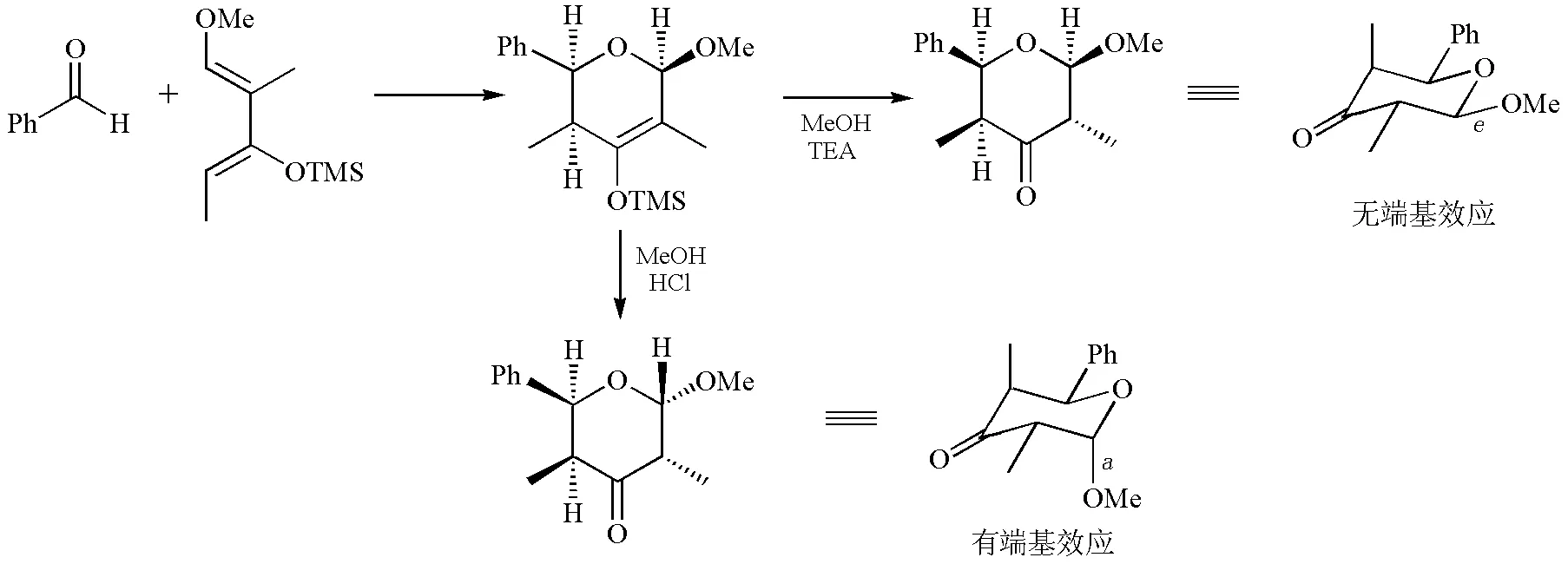
图17 端基效应对烯基硅醚酸性水解的选择性控制
分子内烯烃的氧钯化反应受到端基效应的控制,生成的中间体用CO/甲醇体系捕获,得到以α-端基异构体为主的产物,甲氧基和氢为顺式。作为对比,当甲氧基改为甲基时,在相同的反应条件下,端基效应不可能存在,因此得到的产物没有非对映选择性(图18)[14]。
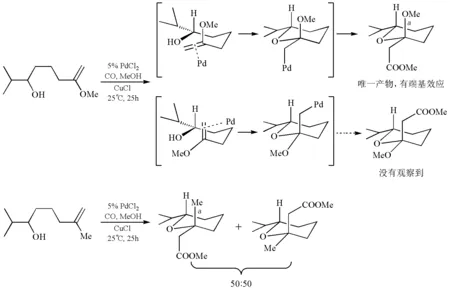
图18 端基效应对氧钯化反应的选择性控制
3.2 应用于有机合成
端基效应控制着产物的立体化学,在有机合成,尤其是全合成领域发挥着重要的作用。如spirastrellolide A的合成,合成的关键是中间体螺缩酮(18)的立体选择性合成。Hsung等巧妙地运用了端基效应,控制了烯烃复分解反应的立体化学[15],合成了有效中间体18(图19)。

图19 端基效应对螺缩酮形成的选择性控制
另一个有效控制螺缩酮立体选择性合成的例子也是通过端基效应实现的[16]。对化合物19实施Pauson-Khand环加成,得到产物20,结构中带两个端基效应,两个氢成反式;当对化合物21实施Pauson-Khand环加成时,为了满足产物22有两个端基效应,两个氢成顺式,此时乙酰氧基(OAc)在直立键上,产物20和22的结构均经过单晶衍射得以证明(图20)。

图20 端基效应对Pauson-Khand环加成的选择性控制
当用R基团(甲基或乙基)代替氢时,端基效应依然存在,如化合物23和25经Pauson-Khand环加成得到立体选择性产物24和26。
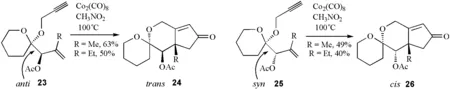
图21 端基效应对Pauson-Khand环加成的选择性控制
化合物22中的乙酰氧基(OAc)占据直立键,当乙酰氧基(OAc)分别用三乙基硅基(TES)、叔丁基二苯基硅基(TBS)和叔丁基二苯基硅基(TBDPS)代替时(图22),随着立体位阻增大,预期的产物28的比例逐渐变小;当取代基是OTBDPS时,以反式的产物29为主,结构中的两个C—O单键均为直立键,没有端基效应,即端基效应被空间效应所克服。

图22 空间效应对Pauson-Khand环加成的选择性控制
4 结论
端基效应是一种很有趣的立体化学现象,虽然不符合取代环己烷的稳定构象的一般规律,但在糖化学中比较常见,而且在有机合成中已得到广泛应用,特别是已被成功地用于不少有机反应的立体选择性控制。用静电排斥作用、偶极矩作用以及分子轨道理论能够从不同的角度对端基效应做出合理的解释。相信本文所介绍的端基效应现象有助于丰富基础有机化学中立体化学、碳水化合物化学等方面的教学内容。
参 考 文 献
[1] 王彦广,吕萍,张殊佳,等.有机化学.第2版,北京:化学工业出版社,2009
[2] 胡宏纹.有机化学.第3版.北京:高等教育出版社,2006
[3] 邢其毅,裴伟伟,徐瑞秋,等.有机化学.第3版.北京:高等教育出版社,2006
[4] McMurry J.Organic Chemistry.7th ed.Singapore:Thomson Brooks/Cole Press,2008
[5] Loudon M G.Organic Chemistry.4th ed.New York:Oxford Univ Press,2002
[6] Lemieux R U,Pavia A A,Martin J C,etal.CanJChem,1969,4427
[7] Palleros D.Carbohydrates.http://www.chem.ucsc.edu/courses/palleros/
[8] Romers C.TopStereochem,1969(4):39
[9] Lee S S,Greig I R,Vocadlo D J,etal.JAmChemSoc,2011,133(40):15826
[10] Giese B,Dupuis J.TetrahedronLett,1984,25:1349
[11] Perron F,Albizati K F.ChemRev,1989,89:1617
[12] Brimble M A,Furkert D P.CurrOrgChem,2003,7:1461
[13] Danishefsky S,Langer M.JOrgChem,1985,50:3674
[14] Semmelhack M F,Kim C,Zhang N,etal.Pure&ApplChem,1990,62(10):2035
[15] Liu J,Hsung R P.OrgLett,2005,7(11):2273
[16] Ghosh S K,Hsung R P,Liu J.JAmChemSoc,2005,127:8260
猜你喜欢
系统仿真技术(2022年4期)2023-01-17
化学工程师(2022年5期)2022-05-11
浙江化工(2022年4期)2022-05-07
光子学报(2022年3期)2022-04-01
贵州大学学报(自然科学版)(2022年1期)2022-01-26
科学之谜(2019年9期)2019-10-16
国外医药(抗生素分册)(2016年4期)2016-07-12
信息记录材料(2016年4期)2016-03-11
合成化学(2015年9期)2016-01-17
烟草科技(2015年8期)2015-12-20
