
硫对InN(0001)面性能的影响
2012-06-06 06:57韦俊红李玉琦张文庆
河南科技学院学报(自然科学版) 2012年1期
韦俊红,李玉琦,张文庆
(河南科技学院,河南新乡453003)
自从Sandroff等人发现通过硫对GaAs(0001)进行简单的化学处理以后[1],GaAs表面的电子性质得到了很大的改善,一系列有关硫在III~V表面钝化的实验和理论研究也引起了人们极大的兴趣,先后有许多学者对GaAs、Si、InAs、Ge、InP、GaN等表面做了有关硫钝化的研究.对于III族N型材料,表面本身就存在着损耗层,有关实验已经证实了硫的钝化减小了表面的重构速率[2].Plucinski用ARPES(angle-resolved photoemission spectroscopy)的方法对GaN低指数面做了硫的钝化研究[3],发现由于硫单层的存在,减少了氧原子在GaN表面的吸附,同时发现表面的费米能级在氧、硫吸附后变小了.也有大量的理论工作者运用第一原理的方法进行模拟计算,如Ferraz、Hirsch等人分别对InP、GaAs的低指数面(0001)面进行了计算[4],通过对比硫在不同的覆盖度下的形成能,得出最稳定的结构是硫的钝化和替换并存的结构.硫钝化InN(0001)面是否能改善表面的电子性质,目前有关研究还未见报道.本文主要研究硫原子钝化对InN(0001)表面性能的影响.
1 模型和方法
基于第一原理赝势方法,采用超原胞模型,分别研究了氧、硫原子在InN(0001)表面的吸附以及硫原子的钝化行为.在计算中采用基于密度泛函理论和局域密度近似的VASP程序包[5],In4d电子作为价电子处理,价电子与离子实的相互作用由US-Vanderbilt超软赝势描述,总能的优化过程用共轭梯度方法,波函数的截断能量为395.7 eV.模型选取为2×2的6个InN双原子层为衬底,表面2个In-N双层以及吸附原子允许弛豫,其他原子不动,见图1(a).计算(0001)面吸附再构时,为避免In-和N-终端之间的电子转移,衬底另一面氮原子的悬挂键用0.75 e赝H饱和,真空层厚度取为1.43 nm.自洽迭代过程中,布里渊区的积分K点选取了Gamma 4×4×1格点.分别计算了氧硫原子在不同覆盖度下、氧在硫钝化前后在3个高对称点的吸附能,并对各种吸附结构的稳定性做了定性的分析.
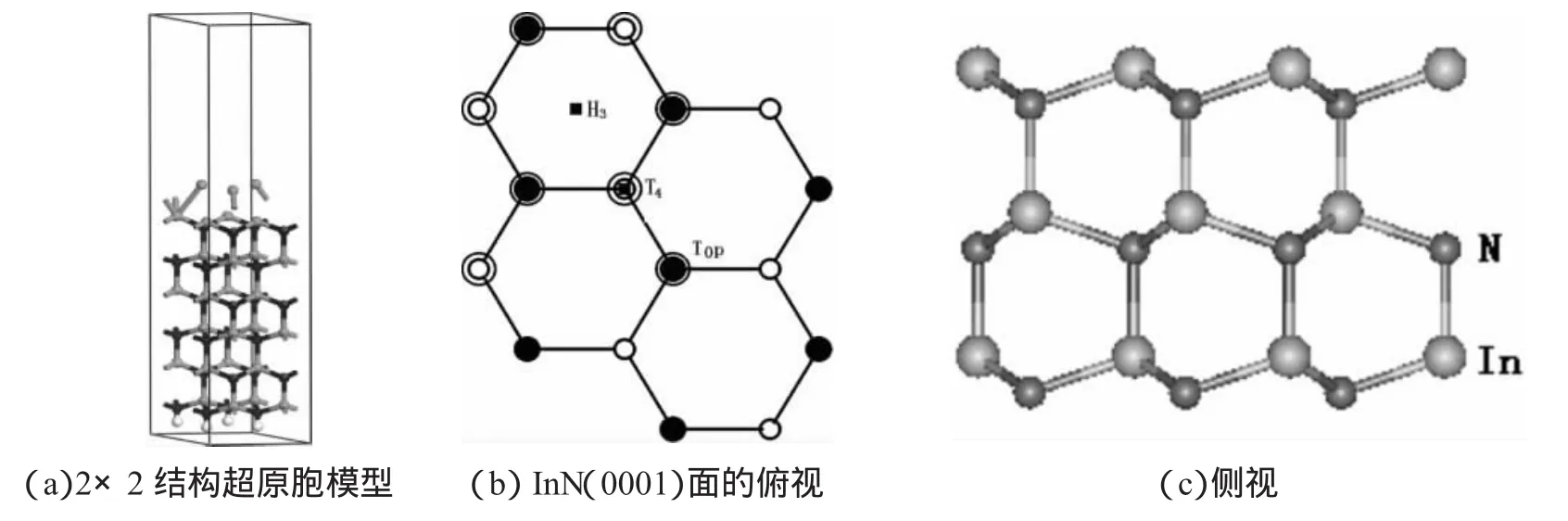
图1 InN体材料结构
在研究InN(0001)表面的吸附结构时,仅考虑了高对称点的吸附和空位结构,如TOP、H3和T4位.如果每个这种位置都被1个铟或氮吸附原子占据,就是l×l结构,研究了1×1上的单个硫﹑氧原子分别在(0001)面 TOP、T4、H3位置的吸附,单个氧原子分别在吸附硫后的 TOP、T4、H3位置的吸附.除了 l×1 结构,我们还研究了2×1、2×2等结构.
为了比较InN(0001)表面的多种表面再构的相对稳定性,不同结构的形成能Eform由以下公式来计算:

式(1)中Ef是所求的能量,Etot表示系统的总能,Ec表示清洁表面弛豫后的能量,i表示吸附原子的种类,是吸附原子的化学势,ni是吸附原子的的数量.
系统中的化学势,它们两个不是独立的,有这样的关系:

lk),即

2 结果分析和讨论
2.1 硫原子对氧在InN(0001)面吸附的影响
首先对InN晶格常数进行了优化,优化后的晶格常数a=0.352 16 nm,c=0.570 1 nm与理论值a=0.353 65 nm,c=0.570 39 nm符合的比较好,在本文的研究中用的均是优化后的值.
2.1.1 氧、硫原子分别在纯净InN(0001)表面不同结构的吸附 能量优化后,通过比较各个不同位置的形成能,氧吸附在H3位的总能比T4、TOP位分别低0.82 eV、1.68 eV,随着覆盖度降低而降低.即覆盖度为3/4、1/2、1/4 时,氧原子在 H3位的吸附能均比较低,比 T4、(TOP)位分别低 1.419(1.758)eV、1.00(1.91)eV、1.212(2.749)eV.从形成能与覆盖度的关系中可以看出,随着氧覆盖度的增加,氧吸附结构的形成能明显地增加,见图2(a).在2×2中H3位的吸附是最稳定的结构,这与已经报道的结论相一致[6].造成这一现象的原因可能是由于氧原子的电负性较高,In-O键的形成使氧成为一个负电荷离子.TOP位能量最高,最不稳定,可能由于TOP位比其他两个高对称位置距离氮原子远,排斥作用较弱的缘故.在实际的生长过程中,有少量的氧存在和表面发生反应使InN体材料中形成杂质,这也是造成InN体材料不纯的原因之一.
对于一层的硫原子在表面的形成能较大,和氧的吸附情况相比,各相应高对称位能量差值不大.其中H3位能量最低,比T4、TOP位能量分别低0.56 eV、0.04 eV.当覆盖度为3/4、1/2、1/4时,能量最低点仍是H3位,比 T4、(TOP)位能量低 1.045(0.62)eV、0.53(0.4)eV、0.467(0.88)eV,T4位能量最高,最不稳定.在图 2(b)中可以清晰看到硫在InN表面的形成能和氧吸附的规律一致,在H3位置的吸附是稳定的,但是各高对称位形成能相比很低,并且没有氧的形成能变化明显,说明硫原子没有氧那么容易在氮化铟(0001)表面成键.可能是由于位于同一主族的氧硫原子,硫的电负性(2.5)没有氧的电负性(3.5)强,硫原子没有氧原子更容易得到电子所致.
2.1.2 硫原子对InN(0001)表面氧吸附的影响 首先在各个不同结构上放单层的硫原子,氧原子在硫原子之上,氧的吸附仅考虑了高对称点.从最后优化的结果来看,硫的掺入完全改变了氧单独在(0001)面吸附的规律,H3位不再是能量的最低点,随着覆盖度的不同得到的能量最低点也各不相同.我们列出了形成能与覆盖度的关系图2(c),可以明显看出,氧在表面的吸附能都很小,最稳定的位置是硫在T4或Top位,这一结果和氧直接在清洁InN表面吸附的稳定位置的形成能相比低2.06 eV,如2(a)图.在这一过程中,由于硫的存在很大程度抑制了氧的吸附,很好地起到保护表面的作用,这个结果与Plucinski等所做的实验结果相吻合[3].

图2 氧、硫原子在InN(0001)面形成能与覆盖度的关系
2.2 硫钝化对InN(0001)面的影响
在研究硫钝化InN(0001)面时,选取了2×1不同的结构进行能量优化:①表面覆盖了整个单层的硫原子(SIn);②表面覆盖整个单层硫的同时一个硫原子替代第二层的氮原子(SInS);③表面覆盖半个单层的硫原子(SN);④表面覆盖半个单层硫的同时一个硫原子替代第二层的氮原子(SNS).如图3所示,1、2 灰色球代表硫原子,3、4、7、8 白色球代表铟原子,5、6 黑色球代表氮原子.图 3(a)表示①时的结构,当表示结构③时表层只有灰色球1,其它原子不变;图3(b)表示②时的结构,当表示结构④时表层只有灰色球1,黑色球6替换为灰色,其它原子不变.
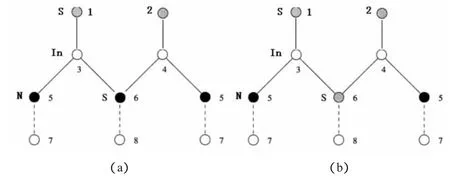
图3 硫钝化InN的不同结构
计算了硫在不同结构钝化的形成能,计算结果表明,SInS比SIn结构形成能低3.188 eV,SN和SNS结构比SIn结构形成能分别高1.56 eV、0.832 eV.SNS比SN结构的形成能低0.728 eV,但是与SInS结构相比能量又高出了4.02 eV.结果对比后得出形成能最低的结构是SInS,即在所有研究结构中,形成能最低的是表面覆盖整个单层硫原子的同时一个硫原子替代第二层的氮原子.造成这一现象的原因可能是第二层替代的硫原子发生扩散从而使结构总能量降低了.这一结果与曾经研究相似模型得到的结果相一致[7].图4是不同结构的形成能与硫的化学势变化关系.
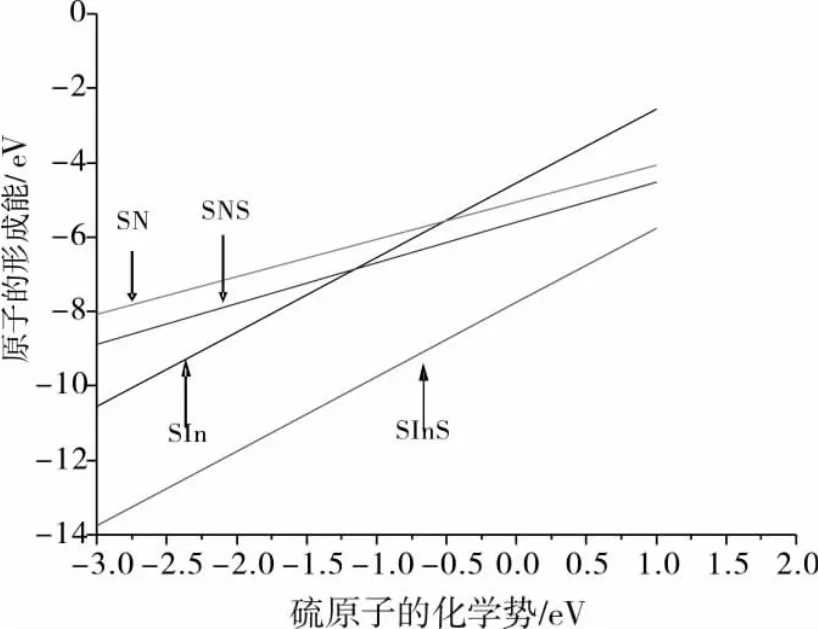
图4 形成能Ef与化学势关系
原子位置由最小总能优化后计算确定,比较了硫钝化前后衬底表面的两个双层和吸附原子驰豫的位置坐标,着重研究竖直方向的坐标.计算结果表明:干净InN(0001)表面驰豫很小,对吸附再构基本没有影响,第一、二双原子层内部扩张,双层与双层间键收缩,衬底两个驰豫双层总体均表现出收缩趋势.SIn结构中,顶部吸附的硫原子和理想位置相比向内收缩了0.004 nm,和单层硫原子(SN结构)相比,尽管都向内收缩了,但是双层硫原子的吸附收缩的程度较大.结构最稳定的SInS结构中,表层两个硫原子均向内收缩,收缩高度大小不同,在替代原子顶部的硫原子向内收缩0.035 4 nm,替代的硫原子向外扩张了0.003 4 nm.说明表面的S-N键没有In-S键稳定,硫原子主要和表面的铟原子成键,硫取代表面的铟原子形成桥键.In-S键的生成对表面的氧化有很强的抑制作用,从而改善了表面特性,降低了表面复合速度.
3 结论
本文用第一性原理计算的方法研究了硫原子对InN(0001)面氧吸附的影响以及硫钝化对InN(0001)面性能的影响.结果表明:硫原子引入后,氧在各个结构高对称位的吸附能与引入前相比,普遍降低,说明硫原子没有氧那么容易在InN(0001)表面成键,很大程度地抑制了氧的吸附,很好地起到保护表面的作用;在硫钝化的过程中,最稳定的SInS结构,表层两个硫原子均向内收缩,顶部的硫原子向内收缩0.035 4 nm,替代氮的硫原子向外扩张了0.003 4 nm.说明表面的S-N键没有In-S键稳定,硫原子主要和表面的In原子成键,硫取代表面的铟原子形成桥键.In-S键的生成对表面的氧化有很强的抑制作用,从而改善了表面特性,降低了表面复合速度.
[1]Sandroff C J,Nottenburg R N,Bischoff J,et al.Dramatic enhancement in the gain of a GaAs/AlGaAs heterostructure bipolar transistor bysurface chemical passivation[J].Applied Physics Letters,1987,51(1):33-35.
[2]CarpenterMS,MellochMR,DunganTE.Schottkybarrierformationon(NH4)2S-treatedn-andp-type(100)GaAs[J].AppliedPhysics Letters,1988,53(1):66-68.
[3]Plucinski L,Colakerol L,Bernardis S,et al.Photoemission studyofsulfur and oxygen adsorption on GaN(0001)[J].Surface Science,2006,600(1):116-123.
[4]Hirsch G,Krüger P,Pollmann J.Surface passivation of GaAs(001)by Sulfur:ab initio studies[J].Surface Science,1998,402-404(15):778-781.
[5]Kresse G,Hanfner J.Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in Germanium[J].Physical Review.B,Condensed Matter,1994,49(20):14251-14269.
[6]Dai X Q,Wang J L,Xie M H,et al.Structural properties of oxygen on InN(0001)surface[J].Surface Science,2007,601(2):161-165.
[7]Qian GX,Martin R M,Chadi DJ.First-principles studyofthe atomic reconstructions and energies ofGa-and As-stabilized GaAs(100)surfaces[J].Physical Review.B,Condensed Matter,1988,38(11):7649-7663.
猜你喜欢
分子催化(2022年1期)2022-11-02
科学技术创新(2022年30期)2022-10-21
农业与技术(2021年23期)2021-12-14
椰城(2021年12期)2021-12-10
黑龙江水利科技(2020年8期)2021-01-21
少儿科学周刊·少年版(2021年22期)2021-01-17
原子与分子物理学报(2020年5期)2020-03-17
电子制作(2019年15期)2019-08-27
测控技术(2018年9期)2018-11-25
物理学报(2017年21期)2017-11-10
