
大气汞形态分布的研究进展
2012-03-30 08:23刘燕罗津晶
环境科学导刊 2012年6期
刘燕,罗津晶
(厦门大学环境科学研究中心,福建厦门361005)
大气汞形态分布的研究进展
刘燕,罗津晶
(厦门大学环境科学研究中心,福建厦门361005)
归纳了大气中汞的形态和行为,并联系近年来国内外对海洋边界层大气汞的研究现状,介绍本实验室对厦门近海地区气态元素汞和颗粒态总汞同步监测的研究,考察厦门近海地区大气汞的分布特征及影响因素,以期补充近海地区汞污染数据,为汞污染控制和治理工作提供数据支持。提出了今后应进一步开展的研究方向。
大气汞;近海地区;形态分布;海洋边界层
重金属汞是构成地壳的物质,在自然界中分布比较广泛。由于它特殊的理化性质,决定了它可以通过沉降机制进入各生态系统,并通过食物链的富集最终对人类及生物的中枢神经系统造成严重的损伤。大气汞有自然来源和人为来源,这两种来源每年向大气环境排放的汞量是极其可观的。目前普遍接受的多种自然来源向大气环境排放的汞量范围是每年1000~4000 t[1],Seigneur估算每年人为汞排放源向大气环境排放的汞有2000~2100 t[2]。高排放量会引起高沉降量,其对生态系统的影响不容忽视。近年来,随着化学燃料燃烧、金属冶炼等人为活动以及对能源的需求加剧,不断向大气环境释放大量的汞,具有特殊物理化学性质的汞已经成为一种通过大气环境进行跨国传输、备受关注的全球性污染物[3]。
1 大气汞的存在形态及行为
大气汞主要有三种价态[4]:气态元素汞GEM(elemental gaseous mercury,Hg0)、活性气态汞RGM(reactive gaseous mercury,主要是Hg2+)、颗粒态总汞TPM(totalparticulatemercury,Hg(p)),而GEM和RGM统称气态总汞TGM(total gaseous mercury),在TGM中GEM所占比例大于95%。研究显示,北半球地区TGM的背景值浓度[5]为1.5~2.0 ng/m3,而其在南半球的背景值浓度[6]为1.1~1.4 ng/m3,北半球地区大气GEM的背景浓度值[7]为1.0~2.0 ng/m3,RGM的背景值[8]是GEM的3%(1~600 pg/m3),大气TPM的背景浓度[8]为1~86 pg/m3。
1.1 大气中各种形态汞的环境行为
大气汞经过物理化学转化后最终沉降至陆地,其种类以及化学形态决定了它们的沉降机制和循环机制。由于GEM具有易挥发性、较低的水溶性以及不稳定性,故GEM在大气中可以进行长距离的迁移,滞留时间可以长达1~2 a[9]。然而RGM和TPM较易溶于水,且具有较高的干沉降速率(RGM:Vd=0.5~6 cm/s;TPM:Vd=0.02~2.0 cm/s)[10~11],因此RGM和TPM在释放源以及污染源附近便可以通过干湿沉降作用进入陆地及水生生态系统[12],在大气中滞留时间通常在几小时到几周,一般不参与大气长距离传输。
1.2 大气中各种形态汞的相互转化
尽管RGM和TPM在大气总汞中所占的比例很小(通常在pg/m3的水平),但是它们对大气汞的沉降和去除过程具有重要的意义。一方面,RGM由于水溶性而易溶解于雨水和云层,TPM除了重力沉降等自然沉降外,易被雨水冲刷而沉降至陆地和水生生态系统,因而TPM和RGM是大气干湿沉降的主要来源;另一方面,三种形态汞在大气中能发生复杂的相互转化(如图1),占TGM 95%以上的GEM在大气中通过光化学反应被强氧化物质(如O3、H2O2、卤素等)氧化为RGM,而RGM和GEM又易被气溶胶吸附形成TPM,因此GEM转变为TPM和RGM是大气汞主要的去除方式。研究显示,在对流层中滞留时间长达几个月至1.5a的GEM,其滞留时间在海洋边界层(Marine Boundary Layer)[13]和极地地区的春季[14]可以缩短至几周甚至几个小时。这是由于在海洋边界层和极地大气中存在大量的活性卤素自由基(Cl、Br、ClO、BrO等),边界层内光化学反应越多,GEM损失则越多[15]。受海洋边界层影响的地区,大气中GEM或TGM的浓度相对内陆等地区较低,有些甚至低于背景浓度值[16~18]。因此,研究海洋边界层内各形态汞的比例以及转化机制,有助于深入了解各形态汞在海陆边界的行为。
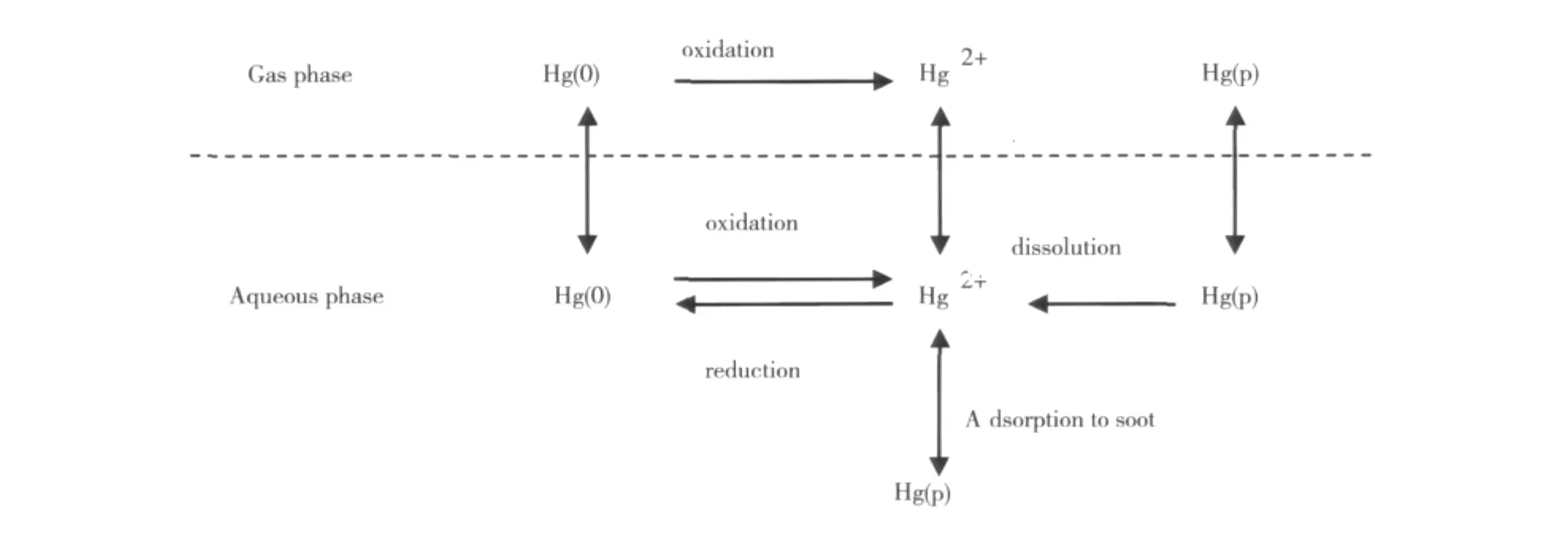
图1 汞在液相和气相中的转化途径[19]

表1 海洋边界层的TGM和GEM监测值统计[27]
2 大气中不同形态汞的研究进展
2.1 不同区域大气形态汞的浓度比值
近年来,全球各地对大气中各形态汞的监测数据日益完善,但是监测点以内陆居多,其次是湖泊、海洋表面,而对近海地区海洋边界层内大气汞的监测较为缺乏。
在我国的山林地区,诸如公戈山、鹿林山等[20~22],这些地区大气中TGM与GEM同RGM、TPM的浓度比值分别是:(1∶0.08%∶0.41%)、(1∶0.7%∶0.13%)。然而,在我国的内陆地区,部分地区大气中气态汞同颗粒态汞的比值相对较大,例如北京[23](1∶17.61%)、重庆[24](1∶16.86%)、吉林[25~26](1∶1.93%)。这些地区大气中TPM浓度相对较高的主要原因是冬季供暖期燃烧煤,杨永奎等人[3]在研究中提到煤中的汞在燃烧过程有75%排放到大气中,这些排放的汞附着在气溶胶中,导致大气中颗粒态汞浓度高于一般水平。
而以往对海洋边界层区域的大气汞研究,主要是对TGM浓度的监测。Kang在其对日本海的海洋边界层TGM浓度的研究中,统计了以往国内外对海洋边界层的TGM或GEM浓度的研究,表1中显示的TGM和GEM的检测值都比较低,均接近背景值(1.5~2.0 ng/m3)。
Kang认为,海洋边界层中存在的氧化物是导致大气中TGM和GEM浓度降低的主要原因,这也是其他学者对海洋边界层内气态汞浓度相对较低的普遍解释。但是若能同一时间采集多种形态的汞,并结合气象等因素将采样区内3种形态的浓度比值同其他非海洋边界层地区的检测值进行对比,能更好地探讨并解释不同形态汞在海洋边界层内的转化机制。因此,研究海洋边界层区域内大气汞形态的监测工作有深刻的研究意义。
2.2 厦门近海地区大气汞浓度分布
厦门地处台湾海峡南部西侧、厦门湾内,属于海湾型城市,盛行偏东风,属亚热带海洋性季风气候,太阳辐射较强。研究厦门近海地区大气中不同形态汞的浓度分布,并考察该地区大气汞的分布特征及影响因素,能够补充近海地区汞形态分布数据,为汞污染控制和治理工作提供数据支持。
2011年2 月~2012年3月,于厦门大学海洋楼楼顶(24°26'08″N,118°05'25″E)设大气汞采样点。采样装置参照Keeler[28]的采样箱设计,通过金管分析法和高温还原法分别分析GEM和TPM样品,最后用冷原子荧光光谱法检测。该采样点距离海岸边仅55 m左右,受海洋边界层的影响较为明显。采样点周围主要的污染源与采样点的相对位置如图2所示,其中漳州某电厂P1距采样点25.8km、厦门某大型燃煤电厂P2距采样点6.9km、厦门某垃圾焚烧电厂P3距采样点11km、厦门某燃煤电厂P4距采样点16km、西部某垃圾焚烧发电厂P5距采样点11.8km。

图2 采样点(A1)周围污染源(P1~P5)点位图
统计得2011年2月~2012年3月,GEM和TPM的浓度范围分别是1.92~12.86 ng/m3、42.96~3620.0 pg/m3,平均浓度值分别是5.54 ng/m3、633.43 pg/m3。从统计学的正态分布角度看,GEM的浓度大致集中在3~8 ng/m3,而TPM的浓度主要集中在<1000 pg/m3的范围。
GEM与TPM的浓度值比例为1∶11.4%,该比例显著高于背景点各形态汞之间的比值,且与内陆地区大气汞各形态间的比值相近。2010年2月~2011年3月,于同样地点采样TPM,结合空气中O3的浓度值,统计得TPM和O3之间呈正相关性,当空气中O3浓度增加时,TPM也呈增加的趋势。Schroeder[29]认为,当空气中的O3浓度增加时GEM浓度降低。除了污染源的直接排放,TPM的增加可能是由空气中的GEM转化而来。
该研究有以下方面工作需要进一步开展:①增加大气中RGM的采集分析,完善大气中3种形态汞的同步监测;②增加采样点的数量,包括在上风向处设置参考点、在下风向处增设置2~3处采样点;③结合采样期间的气象参数(如风向、风速、太阳辐射量、相对湿度)、污染物浓度(如O3、SO2等),综合分析各形态汞与各影响因素的相关性。
3 小结
大气汞在环境中主要有3种形态,并且在一定条件下能发生相互转化。大气汞通过干湿沉降进入陆地和水生生态系统,并最终由食物链对人类和生物产生毒害作用,是一种能进行跨国传输的全球性污染物。因此,正确地了解汞在大气环境中的形态和行为是一项十分迫切的任务,只有全面了解汞的行为及浓度分布特征,才能正确地评估大气汞的形态转化、迁移模式等,才能采取合适的手段从根本上控制汞的排放。本实验室对厦门近海地区气态元素汞和颗粒态总汞进行同步监测,考察该地区大气汞的分布特征及影响因素,以补充近海地区汞污染数据,为汞污染控制和治理工作提供数据支持。但是,目前该研究还有多方面的工作需要补充和完善。
[1]冯新斌,仇广乐,付学吾,等.环境汞污染[J].化学进展,2009,21(2):436-458.
[2]Seigneur C,Vijayaraghavan K,Lohman K,et al.Global source attribution for mercury deposition in the United States[J].Environmental Science&Technology,2004,38(2):555-569.
[3]杨永奎,王定勇.大气汞的时空分布研究进展[J].四川环境,2007,25(6):91-95.
[4]Pleijel K,Munthe J.Modeling the atmospheric chemistry of mercury[J].Water,Air,&Soil Pollution,1995,80(1):317-324.
[5]Lamborg C H,Fitzgerald W F,O'Donnell J,et al.A nonsteady-state compartmental model of global-scale mercury biogeochemistry with interhemispheric atmospheric gradients[J].Geochimicaet Cosmochimica Acta,2002,66(7):1105-1118.
[6]Baker P,Brunke E G,Slemr F,et al.Atmospheric mercury measurements at Cape Point,South Africa[J].Atmospheric Environment,2002,36(14):2459-2465.
[7]Landis M S,Keeler G J,Al-Wali K I,et al.Divalent inorganic reactive gaseous mercury emissions from a mercury cell chlor-alkali plant and its impact on near-field atmospheric dry deposition[J].Atmospheric Environment,2004,38(4):613-622.
[8]Keeler G,Glinsorn G,Pirrone N.Particulate mercury in the atmosphere:its significance,transport,transformation and sources[J].Water,Air,&Soil Pollution,1995,80(1):159-168.
[9]Lin C J,Pehkonen S O.The chemistry of atmospheric mercury:a review[J].Atmospheric Environment,1999,33(13):2067-2079.
[10]Seth N,Gustin M S,Prestbo E M,et al.Estimation of dry deposition of atmospheric mercury in Nevada by direct and indirect methods[J].Environmental Science&Technology,2007,41(6):1970-1976.
[11]Poissant L,Pilote M,Xu X,et al.Atmospheric mercury speciation and deposition in the Bay St.Francois wetlands[J].J.Geophys.Res,2004,109(11).
[12]Schroeder W H,Munthe J.Atmospheric mercury-an overview[J].Atmospheric Environment,1998,32(5):809-822.
[13]Hedgecock I M,Pirrone N.Chasing quicksilver:Modeling the atmospheric lifetime of Hg0(g)in the marine boundary layer at various latitudes[J].Environmental Science&Technology,2004,38(1):69-76.
[14]Skov H,Christensen J H,Goodsite M E,et al.Fate of elemental mercury in the Arctic during atmospheric mercury depletion episodes and the load of atmospheric mercury to the Arctic[J].Environmental Science&Technology,2004,38(8):2373-2382.
[15]Weiss-Penzias P,Jaffe D A,McClintick A,et al.Gaseous elemental mercury in the marine boundary layer:Evidence for rapid removal in anthropogenic pollution[J].Environmental Science&Technology,2003,37(17):3755-3763.
[16]Ci Z,Zhang X,Wang Z,et al.Atmospheric gaseous elemental mercury(GEM)over a coastal/rural site downwind of East China:temporal variation and long-range transport[J].Atmospheric Environment,2011:2480-2487.
[17]Chand D,Jaffe D,Prestbo E,et al.Reactive and particulate mercury in the Asian marine boundary layer[J].Atmospheric Environment,2008,42(34):7988-7996.
[18]Laurier F J G,Mason R P,Whalin L,et al.Reactive gaseous mercury formation in the North Pacific Ocean’s marine boundary layer:A potential role of halogen chemistry[J].J.Geophys.Res,2003,108(D17):4529.
[19]Ryaboshapko A,Bullock R,Ebinghaus R,et al.Comparison of mercury chemistry models[J].Atmospheric Environment,2002,36(24):3881-3898.
[20]Fu X,Feng X,Wang S.Exchange fluxes of Hg between surfaces and atmosphere in the eastern flank of Mount Gongga,Sichuan province,southwestern China[J].Journal of Geophysical Research,2008,113(D20):1-12.
[21]Fu X,Feng X,Zhu W,et al.Total particulate and reactive gaseous mercury in ambient air on the eastern slope of the Mt.Gongga area,China[J].Applied Geochemistry,2008,23(3):408-418.
[22]Sheu G R,Lin N H,Wang J L,et al.Temporal distribution and potential sources of atmospheric mercury measured at a highelevation background station in Taiwan[J].Atmospheric Environment,2010,44(20):2393-2400.
[23]Wang Z,Zhang X,Chen Z,et al.Mercury concentrations in size-fractionated airborne particles at urban and suburban sites in Beijing,China[J].Atmospheric Environment,2006,40(12):2194-2201.
[24]王定勇.重庆大气汞初步调查[J].重庆环境科学,1996,18(4):59-62.
[25]Wan Q,Feng X,Lu J,et al.Atmospheric mercury in Changbai Mountain area,northeastern China I.The seasonal distribution pattern of total gaseous mercury and its potential sources[J].Environmental research,2009,109(3):201-206.
[26]Wan Q,Feng X,Lu J,et al.Atmospheric mercury in Changbai Mountain area,northeastern China II.The distribution of reactive gaseous mercury and particulate mercury and mercury deposition fluxes[J].Environmental research,2009,109(6):721-727.
[27]Kang H,Zhouqing X.Atmospheric mercury over the marine boundary layer observed during the third China Arctic Research Expedition[J].Journal of Environmental Sciences,2011,23(9):1424-1430.
[28]Keeler G J,Landis M S.Standard Operating Procedure for Analysis of Particulate Phase Mercury[J].Ann Arbor,1994,1001:48109-42029.
[29]Schroeder W H,Yarwood G,Niki H.Transformation processes involving mercury species in the atmosphere—results from a literature survey[J].Water,Air,&Soil Pollution,1991,56(1):653-666.
The Research Progress on Atmospheric Mercury Distribution
LIU Yan,LUO Jin-jing
(Environmental Science Research Center,Xiamen University,Fujian Xiamen 361005 China)
This paper summarizes the morphology and behavior of the atmospheric mercury,including the current research of the atmospheric mercury in the marine boundary layer.It also introduces the synchronized monitoring and research of the elemental gaseous mercury and total particle mercury done by our laboratory in the offshore area of Xiamen,where the distribution characteristics of the atmospheric mercury and its influence factors are studied.The research is expected to provide the data about offshore mercury pollution,in order to support the mercury pollution control.
atmospheric mercury;offshore area;morphological distribution;marine boundary layer
X16
A
1673-9655(2012)06-0009-04
2012-04-25
福建省重点科技项目(No.2008I0024)。
刘燕,硕士研究生,研究方向:大气汞监测。
猜你喜欢
中国自行车(2022年6期)2022-10-29
航空发动机(2020年3期)2020-07-24
中国特种设备安全(2020年11期)2020-06-09
海峡姐妹(2018年12期)2018-12-23
柴油机设计与制造(2018年3期)2018-10-13
柴油机设计与制造(2018年2期)2018-08-29
柴油机设计与制造(2018年1期)2018-04-20
海峡姐妹(2017年6期)2017-06-24
商业文化(2017年23期)2017-04-23
应用数学与计算数学学报(2015年1期)2015-07-20
