
Engineering of Corynebacterium glutamicum to Enhance L-ornithine Production by Gene Knockout and Comparative Proteomic Analysis*
2012-02-14 08:26LUDongmei卢冬梅LIUJianzhong刘建忠andMAOZongwan毛宗万
LU Dongmei (卢冬梅), LIU Jianzhong (刘建忠)** and MAO Zongwan (毛宗万)
The Key Laboratory of Gene Engineering of Ministry of Education, State Key Laboratory of Biocontrol, Sun Yat-Sen University, Guangzhou 510275, China
1 INTRODUCTION
L-ornithine is a non-essential amino acid and an important constituent of the urea cycle. It is also the precursor of other amino acids such as citrulline and arginine. It is effective in the treatments of liver diseases and liver-protecting [1]. It was also applied to wound healing [2]. Recently, it was demonstrated that ornithine supplementation increased growth hormone and insulin-like growth factor-1 serum levels after heavy-resistance exercise in strength-trained athletes[3]. Many studies have reported that L-ornithine was produced from a citrulline- or arginine-required mutant of a coryneform bacterium obtained by classical mutagenesis [4-6]. Although this mutant can produce a high yield of L-ornithine, its growth culture is always unstable because of reversion of the auxotrophic mutant,and then the production of L-ornithine drops markedly.
Recently, metabolic engineering has become a powerful approach for strain improving. It may also be used to enhance the flux to L-ornithine. The common strategy of metabolic engineering is to improve the availability of precursor by knockouts and overexpressions of genes. In Corynebacterium glutamicum,ornithine is synthesized from glutamate through the acetyl cycle of arginine biosynthesis [7]. Only three papers reported the results of enhance of L-ornithine production by metabolic engineering so far [8-10]. Lee and Cho reported that an engineered Escherichia coli W3110 (ΔargFΔargIΔargRΔproBΔspeF, ParaB-arg214)produced L-ornithine of 13.2 mg·g-1(based on dry cell mass) and that addition of glutamate to the culture was favorable for ornithine production in E. coli [8].Hwang et al. also reported that cooverexpression of argCJBD in a triple gene knockout C. glutamicum(ΔargFΔargRΔproB) resulted in L-ornithine production of 16.49 mg·g-1(based on dry cell mass), L-ornithine concentration of 179.14 mg·L-1[9]. They also found that the supply of exogenous glutamate to the culture or the increased availability of endogenous glutamate by overexpression of the pyruvate carboxylase along with inactivation of the phosphoenolpyruvate carboxykinase did not enhance ornithine production in the triple gene (argF, argR and proB) knockout strain of C. glutamicum [9]. It has been reported that proline could be converted into L-ornithine by ornithine cyclodeaminase (Ocd) and Ocd was a key enzyme for enhancing L-ornithine production under prolinesupplemented condition in C. glutamicum [10]. Recent studies have demonstrated that a reduction in the 2-oxoglutarate dehydrogenase complex activity is important for glutamate overproduction by C. glutamicum [11, 12]. Asakura et al. [11] reported that deletion of the odhA gene led to an increase in glutamate production. Kim et al. [12] reported that the inactivation of ODHC by odhA antisense RNA expression could not improved non-triggered-glutamate production, but it could enhance Tween-40-triggered-glutamate production. Thus, the effect of inactivation of ODHC on ornithine production in C. glutamicum was investigated.
Proteomics is playing an important role not only in biological research but also in various biotechnological applications because most cellular metabolic activities are directly or indirectly mediated by proteins. Comparative proteome analysis under various genetic or environmental conditions is carried out, one can identify protein spots that show altered intensities for further analysis and manipulation [13, 14]. Plenty of such studies have been carried out, which resulted in the successful design of strategies for the enhanced production of valuable compounds [13, 15-18]. Thus,comparative proteome analysis between engineered and wild-typeC.glutamicumwas carried out to investigate the mechanism of ornithine production of engineeredC.glutamicum.
2 MATERIALS AND METHODS
2.1 Microorganism and medium
All bacterial strains used in this study are listed in Table 1.E.coliDH5α was used for plasmid construction.C.glutamicumATCC13032 was used as a wild-type strain for the construction of mutant strain.
For the recombinant DNA techniques, Luria-Bertani (LB) was used forE.coliandC.glutamicumas complex medium. For L-ornithine production ofC.glutamicum, the seed medium consisted of (per liter)25 g of glucose, 10 g of yeast extract, 10 g of corn steep liquor, 15 g of (NH4)2SO4, 2.5 g of MgSO4⋅7H2O, 1 g of KH2PO4, 0.5 g of K2HPO4, 0.5 g of Na2HPO4and 10 g of CaCO3. The fermentation medium consisted of (per liter) 100 g of glucose, 20 g of corn steep liquor, 50 g of (NH4)2SO4, 2.5 g of MgSO4·7H2O, 1 g of KH2PO4, 0.5 g of K2HPO4, 0.5 g of Na2HPO4, 20 mg of FeSO4·7H2O, 20 mg of MnSO4⋅4H2O, 2 g of molasses, 1 ml of Tween 80, and 10 g of CaCO3. Initial pH of all the above mediums was adjusted to 7.0.
2.2 Cultivation
For L-ornithine fermentations, a 1.0-ml sample of the seed culture grown at 150 r·min-1and 30 °C for 12 hours was inoculated into 10 ml of the fermentation medium in a 100-ml flask and incubated at 30 °C and 150 r·min-1for 72 hours. When necessary, kanamycin(50 μg·ml-1for E. coli and 25 μg·ml-1for C. glutamicum),chloroamphenicol (20 μg·ml-1for E. coli and 10 μg·ml-1for C. glutamicum), or ampicillin (50 μg·ml-1for E. coli)were added to the medium.

Table 1 Bacterial strains plasmids and primers used in this study
2.3 Primers and gene knockout
All plasmids constructed and primers for this study are listed in Table 1. Chromosomal DNA of C.glutamicum was isolated as described by Eikmanns et al [20]. The preparation of competent cells and electroporation for C. glutamicum was performed as described by Van de Rest et al [21]. The correct mutants of C. glutamicum were confirmed by PCR and checked for the expected phenotype.
The disruption of gene was performed using the non-replicable integration vector pK18mobsacB which allows for marker-free deletion of the target gene [19]. The argF gene (960 bp) was amplified from the genome DNA of C. glutamicum by PCR using primers argF-F and argF-R, and ligated into pMD18-T.The above plasmid was cleaved with Nco I and then religated. The resulting plasmid was then linearized with BamH I and Sph I. The above fragment of target gene containing an internal deletion was inserted into the corresponding site of pK18mobsacB integration vector to obtain the nonreplicable integration vector pKB001. The proB gene (1734 bp) was amplified from the genome DNA of C. glutamicum by PCR using primers proB-F and proB-R. The PCR product was ligated into the Xba I and Hind III sites of pK18mobsacB. The above plasmid was cleaved with PstI and then religated to obtain the nonreplicable integration vector pKB002. The kgd gene (3794 bp) was amplified from the genome DNA of C. glutamicum by PCR using primers kgd-F and kgd-R. The PCR fragment was ligated into pMD18-T. The above plasmid was cleaved with Bgl II and then religated. The resulting plasmid was then linearized with Sma I and Sal I, and the fragment of target gene containing an internal deletion was inserted into the corresponding site of pK18mobsacB integration vector to obtain the nonreplicable integration vector pKB003. Above nonreplicable integration vectors including pKB001,pKB002 and pKB003 were transferred into C. glutamicum to disrupt the site-specific gene using the protocol described by Schäfer et al. [19], respectively.
2.4 RT-PCR
Total RNA from C. glutamicum cells grown for 72 h was isolated with a RNA extraction kit (Dongsheng Biotech, Guangzhou, China) as indicated by the manufacturer. A one-step real-time quantitative RT-PCR(qRT-PCR) was performed with iCycler iQ5 Real Time PCR system (Bio-Rad Laboratories, USA) and the All-in-OneTMqPCR Mix kit (GeneCopoeia, Guangzhou, China). The experiment was carried out with 100 ng of total RNA as the template. PCR conditions were: 50 °C for 30 min and then 95 °C for 10 min,followed by 40 cycles of denaturation at 95 °C for 15 s, annealing at 55 °C for 20 s, and extension at 72 °C for 15 s. To standardize the results, the relative abundance of 16S rRNA was used as the internal standard.
2.5 Acetylglutamate kinase activity assay
C. glutamicum cells were grown in the fermentation medium at 30 °C and 150 r·min-1for 60 hours,harvested by centrifugation during the exponential phase, and washed with 100 mmol·L-1Tris/HCl buffer(pH 7.5). Cells were sonically disrupted. Cell debris was removed by centrifugation at 5000×g and 4 °C for 15 min. The resulting crude extracts were used for enzymatic measurement. Acetylglutamate kinase activity was determined by the ferric chloride method [22, 23].The reaction mixture contained 50 μmol of Tris/HCl buffer (pH 8.0), 20 μmol of sodium N-acetylglutamate,5 μmol each of adenosine triphosphate (ATP) and MgCl2,and 200 μmol of NH2OH⋅HCl (neutralized with KOH)in a total volume of 0.5 ml. After incubation for 30 min at 37 °C, 0.5 ml of 10% trichloroacetic acid and 1.0 ml of 5% FeCl3·6H2O in 1 mol·L-1HCl were added.The mixture was centrifuged, and the optical density of the supernatant fluid at 540 nm was measured. The molar absorption coefficient of this complex under the given conditions was 456 L·mol-1·cm-1. One unit of acetylglutamate kinase was defined as the amount of enzyme which catalyzes the formation of 1 μmol of product per min under assay conditions.
2.6 2-DE image analysis
C. glutamicum was cultivated aerobically on a rotary shaker (150 r·min-1) at 30 °C for 72 h. The cells were harvested by centrifugation, washed twice with saline and resuspended in TE buffer (10 mmol·L-1Tris-HCl, pH 8.0, 1 mmol·L-1EDTA) containing protease inhibitor (Roche Diagnostics, Mannheim, Germany), and disrupted by the Sonic Dismembrator(Fisher scientific, USA). Intact cells and cell debris were removed by centrifugation at 5000×g and 4 °C for 15 min. Prior to solubilization, proteins were precipitated over night with 9 volumes acetone at -20 °C.Protein concentrations were determined by the Bradford method [24]. Proteome analysis of the soluble protein fraction was performed by 2-D gel electrophoresis [25]. 300 μg protein was solubilized in 350 μl solubilization buffer [9 mol·L-1urea, 4% CHAPS, 1%DTT, 1% Bio-Lyte 4/6 and Bio-Lyte 5/7(1∶1)] by vigorous shaking for 1 h at room temperature. The protein samples were loaded on the IPG strips (17 cm,pH4-7, Bio-Rad Laboratories, USA) followed by the rehydration at 20 °C for 12 h on the PROTEAN IEF cell (Bio-Rad Laboratories, USA). The rehydrated strips were focused at 20 °C for 0.1 h at 60 V, 1 h at 150 V, 2 h at 500 V and 1 h at 1000 V followed by a linear increase to 3500 V until a total of 64600 V·h was reached. The second dimension run was carried out using 12% polyacrylamide linear gradient gels in the Protean II apparatus (Bio-Rad Laboratories, USA).Electrophoresis was performed at a maximum voltage of 10 mA per gel until the marker dye had moved out from the IPG gel strip completely. Subsequently, electrophoresis was continued at 20-30 mA per gel until the marker dye reached the end of the gel.
2.7 Gel staining, spot quantification and protein identification
For silver staining, proteins were fixed in fixing solution (10% glacial acetic acid, 40% ethanol) for 1 h.After washing the gel twice (20 min everytime) in 30% (by volume) ethanol, the sensitizing step was carried out by incubation in 0.02% sodium thiosulfate for 1 min. After washing the gel three times (20 second everytime) in distilled water, the silver reaction was performed with a 0.2% silver nitrate solution containing 0.02% (by volume) formaldehyde (37%) for 20 min. After washing the gel three times (20 second everytime) in distilled water, protein spots were developed in the solution [3% sodium carbonate, 0.05%formaldehyde (37%) and 0.0005% sodium thiosulfate].Development of spots was stopped with 0.5% glycine.The stained gels were scanned with ArtixScan F1 (Microtek, China). Platinum software 5.0 (GE Healthcare,USA) was used to identify spots, to match gels, and to quantify spot densities on a volume basis (i.e., integration of spot optical intensity over the spot area). Protein spots with altered protein abundance were analyzed by peptide mass fingerprinting or identified using the master 2-D-gel map of the soluble protein fraction ofC.glutamicum[25, 26]. Protein spots of interest were excised from the stained gels with Pasteur pipettes and subjected to in-gel digestion with trypsin essentially as described previously [25]. MALDI-TOF-TOF mass spectra were obtained on an Ultraflex III (Bruke Daltonics).
2.8 Analysis
Cell growth was monitored by measuring the optical density of the culture at 600 nm (OD600) using a spectrophotometer (Shimadzu Corporation, Japan)after dilution of the culture with 0.2 mol·L-1HCl to dissolve CaCO3. L-ornithine concentrations were determined by the colorimetric method with ninhydrin as described previously [27].
3 RESULTS AND DISCUSSION
3.1 Metabolic engineering of C. glutamicum for L-ornithine production
To construct a host strain capable of accumulating L-ornithine, it was firstly focused on preventing the conversion of L-ornithine into citrilline inC.glutamicum. Thus, theargFgene encoding ornithine carbamoyltransferase was disrupted as described in Material and Methods. The resulting strain was grown in the fermentation medium and L-ornithine production was determined. As shown in Table 2, the knockout of theargFgene increases L-ornithine concentration to 2.13 g·L-1from 0.24 g·L-1of the wild-type strain and L-ornithine content to 0.16 g·g-1from 0.02 g·g-1(based on dry cell mass) of the wild-type strain.In order to prevent glutamate to proline, theproBgene in theargFknockoutC.glutamicumwas disrupted.From Table 2, it is found that the double knockout strain produces L-ornithine concentration up to 2.68 g·L-1and L-ornithine content to 0.21 g·g-1(based on dry cell mass). Similar results were obtained inE.coli[8] andC.glutamicum[9]. The knockouts of theargFandproBgenes enhanced L-ornithine production inE.coli[8] andC.glutamicum[9]. It is also found that the level of L-ornithine production, even if no Tween 80-addition condition (3.09 g·L-1), is higher than that reported by Hwanget al[9]. It may result from by fermentation medium. By comparing these medium,corn steep liquor and molasses may be more suitable nitrogen source than yeast extract for ornithine production. From Table 2, we can also find that the knockouts of genes have a slight positive effect on growth. The similar results were observed by Hwanget al. [9].The knockout ofargFgene improved cell growth significantly.
② Data represent the mean of the triplicate cultures ± standard deviation.
③ The difference was significant atP>0.05 by Tukey’s test.
3.2 Effect of inactivation of ODHC on L-ornithine production
In order to investigate the effect of inactivation of ODHC on L-ornithine production inC.glutamicum,thekgdgene of the above double knockout strain ofC.glutamicumwas further disrupted. The triple knockout strain was grown in the fermentation medium and L-ornithine production was determined. Table 2 shows that the knockout of thekgdgene produces L-ornithine up to 4.78 g·L-1from 2.68 g·L-1of the double knockout strainC.glutamicumATCC13032 (ΔargFΔproB).ODHC catalyzes the oxidative decarboxylation of 2-oxoglutarate to succinyl coenzyme A, which is located at the branch point between the tricarboxylic acid (TCA) cycle and the glutamate biosynthesis pathway. ODHC ofC.glutamicumcontains three subunits including alpha-ketoglutarate decarboxylase(2-oxoglutarate dehydrogenase E1 component,EC:1.2.4.2) coded bykgd(odhA), dihydrolipoamide acetyltransferase (EC:2.3.1.12) coded byaceFand dihydrolipoamide dehydrogenase (EC:1.8.1.4) coded bylpd(found from the genome sequencing) [28].Asakuraet al. demonstrated that deletion of theodhAgene led to an increase in glutamate production inC.glutamicum[11]. It has been reported that ODHC activity was dramatically decreased under glutamate-producing conditions. Biotin limitation, surfactants addition or penicillin addition resulted in a reduction of ODHC activity, then improving glutamate production inC.glutamicum[29, 30]. Deletion of theodhAgene could further enhance biotin-limited, Tween-40-triggered or penicillin-triggered glutamate production byC.glutamicum[11]. The inactivation of ODHC byodhAantisense RNA expression can also improve Tween-40-triggered glutamate production byC.glutamicum[12].Thus, the reason of increase in L-ornithine production may be that knockout of thekgdgene together with Tween-80-triggering improves the availability of precursor (endogenous glutamate). Similar result was obtained inE.coli[8]. Their result showed that the supply of exogenous glutamate to the culture enhanced ornithine production inE.coli[8]. However, the group obtained an opposite result inC.glutamicum. They found that the supply of exogenous glutamate to the culture or the increased availability of endogenous glutamate by overexpression of the pyruvate carboxylase along with inactivation of the phosphoenolpyruvate carboxykinase did not enhance ornithine production in the triple gene (argF,argRandproB) knockout strain ofC.glutamicum[9]. The difference between their and our results may be due to the different genetic backgrounds of strains and triggering conditions.In our study, ornithine production was triggered by Tween 80. However, in the study of Hwanget al., the triple gene (argF,argRandproB) knockout strain ofC.glutamicumproduced ornithine without any triggers [9].In addition, it was found thatC.glutamicumATCC13032(ΔargFΔproBΔkgd) without Tween 80 addition produced 3.09 g·L-1of L-ornithine, which is lower than that (4.78 g·L-1) with Tween 80 addition. This suggests that the deletion of thekgdgene further enhance L-ornithine production in the presence of Tween 80 byC.glutamicumATCC13032 (ΔargFΔproBΔkgd).

Figure 1 Representative 2-DE images of C. glutamicum ATCC 13032 (left), C. glutamicum ATCC 13032 (ΔargFΔproBΔkgd)(right)
As shown in Table 2, it is clear that L-ornithine production (2.50 g·L-1) ofC.glutamicumATCC13032(ΔargFΔkgd) was lower than that ofC.glutamicumATCC13032 (ΔargFΔproB) (2.68 g·L-1). The reason may be that some endogenous glutamate inC.glutamicumATCC13032 (ΔargFΔkgd) was converted into L-proline, thus decreasing L-ornithine production.
In order to investigate the gene expression in different knockout strains, the expression levels of theargBgene were analyzed by RT-PCR. The results were presented in Table 2. As shown in Table 2, the expression levels of theargBgene in the multiple gene knockouts is higher than that in the single gene knockout and the triple gene knockout shows the highest expression level. It is also found that the expression level of theargBgene inC.glutamicumATCC13032(ΔargFΔkgd) is lower than that inC.glutamicumATCC13032 (ΔargFΔproB). The ArgB activity ofC.glutamicumATCC13032 derivatives was also determined(Table 2). Similar results to the expression levels were obtained. The triple gene knockout strain had the highest activity. The ArgB activity ofC.glutamicumATCC13032 (ΔargFΔkgd) was higher than that ofC.glutamicumATCC13032 (ΔargFΔproB). From above results, the trend of ornithine production in the different knockout strain is coincident with that of the expression level of theargBgene and that of Arg activity.
3.3 Comparative proteome analysis
In order to understand the physiological changes at the protein level, the comparative proteomic analysis between the wild-type and the engineered strain was performed using the cells when more ornithine was present (early stationary phase). The representative 2-DE images of the proteomes were presented in Fig. 1.The expression levels of 202 proteins varied significantly inC.glutamicumATCC13032 (ΔargFΔproBΔkgd)compared with those in the wild-type strain. Of these proteins, 52 proteins were identified.
The expression levels of proteins involved in L-ornithine biosynthesis are presented in Fig. 2. As shown in Fig. 2, proteins involved in glycolysis (Pfk:1.48-fold, Fda: 1.65-fold, Tpi: 1.67-fold, Gap: 1.54-fold,Eno: 4.87-fold, AceE: 1.67-fold), TCA cycle (Acn:1.76-fold), glutamate biosynthesis (Gdh: 1.67-fold)and ornithine biosynthesis (ArgJ: 15.69-fold, ArgB:32.75-fold, ArgC: 10.56-fold) ofC.glutamicumATCC13032 (ΔargFΔproBΔkgd) are up-regulated compared with those ofC.glutamicumATCC13032. The up-regulated expression levels of these proteins are beneficial to L-ornithine production in the strain. Enzymes involved in pentose phosphate pathway (HMP)(DevB: 0.80-fold, Gnd: 0.83-fold, Rpe: 0.76-fold, Rpi:0.56-fold), TCA cycle (SdhA: 0.59-fold, SdhB: 0.50-fold,FumC: 0.80-fold, Mqo: 0.55-fold), glutamine biosynthesis (GlnA: 0.80-fold), histidine biosynthesis (HisC:0.77-fold) and pyruvate metabolism (Pox: 0.50-fold,AckA: 0.47-fold, Ldh: 0.18-fold) are down-regulated.The down-regulations of these enzymes promote the flux to L-ornithine and enhance L-ornithine production. Enzymes of serine biosynthesis (SerA: 1.65-fold),alanine biosynthesis (IlvB: 1.60-fold) and L-ornithine metabolism (SpeE: 5.57-fold) are up-regulated. They drive the flux away from L-ornithine and result in decrease in L-ornithine production. It is also found that enzymes involved in translation of macromolecule synthesis (Efg: 0.11-fold, Tuf: 0.21-fold), DtsR(0.24-fold), MalQ (0.18-fold) and degradation of RNA(Pnp: 0.06-fold) are significantly down-regulated. Akr(5.14-fold), OrnA (4.89-fold) and isoprenoid pathway(IspH: 4.82-fold) are highly expressed. InC.glutamicumATCC13032 (ΔargFΔproBΔkgd), ArgF and ProB are not observed.
The up-regulations of expression levels of enzymes involved in glycolysis, glutamate biosynthesis and ornithine biosynthesis, and down-regulations of proteins involved in HMP, pyruvate metabolism,downstream of KG in TCA cycle, glutamine biosynthesis, proline biosynthesis and arginine biosynthesis promote the flux into ornithine, and then resulte in enhancing ornithine production in the engineeredC.glutamicum. The expression levels of enzymes in ornithine biosynthesis (ArgCJBD) are much higher than that in the upstream pathway of glutamate(house-keeping gene proteins). It indicates that overexpression of the genes in the upstream pathway of glutamate to increase the availability of glutamate may further increase L-ornithine production in the engineeredC.glutamicumand the ornithine synthesis enzymes (ArgCJBD) may not be the limiting enzymes in the engineeredC.glutamicum. This strategy has been successfully applied for engineeringE.colito enhance L-valine production [31]. Their comparative transcriptomic analyses between the wild-type and the recombinantE.colishowed that the increase extents of expression levels of theilvCEDgenes was much less than those of theilvBNgenes, and then cooverexpression of theilvBNgenes resulted in L-valine production up to 3.43 g·L-1from 1.31 g·L-1.
The metabolic flux analysis demonstrated that the flux into the HMP decreased and the EMP/HMP ratio was estimated to be 80/20 during glutamate overproduction ofC.glutamicum[32-34]. Thus,down-regulations of proteins involved in HMP indicated that more glutamate was produced by the engineered strain, resulting in an increase in ornithine production.
The up-regulations of expression levels of enzymes related to serine biosynthesis, alanine biosynthesis and L-ornithine metabolism resulted in the flux away from L-ornithine and consequently decreased L-ornithine production. Thus, knockouts of these genes (serA,ilvBandspeE) ofC.glutamicummay be beneficial for improving L-ornithine production. Lee and Cho reported that the inactivation of Spe ofE.coliresulted in 33% increase in L-ornithine production [8].
The down-regulation of Pnp may affect cell growth. It was reported that overexpression of thepnpgene dramatically increased the growth rate ofStreptomyces antibioticus[35]. MalQ is related to maltose utilization. The maltose metabolization pathway ofC.glutamicumhas been proposed, which composes of maltodextrin and glucose formation by MalQ with maltose as substrate, glucose phosphorylation by Glk and maltodextrin degradation via the reactions of maltodextrin phosphorylase (malP) and α-phosphoglucomutase (proBably encoded by cg2523)[36, 37]. Moreover, MalQ is the key enzyme of the maltose metabolization pathway. The significant down-regulation of MalQ indicates that the engineeredC.glutamicumcould not utilize maltose as the sole carbon source. The significant up-regulation of IspH of isoprenoid pathway shows that the engineeredC.glutamicumrequires more oxygen thanC.glutamicumATCC 13032. Thus, supply of dissolved oxygen may be a key fact for L-ornithine production by the engineeredC.glutamicum. The above strategies for engineeringC.glutamicumto improve L-ornithine production are being investigated in our lab.
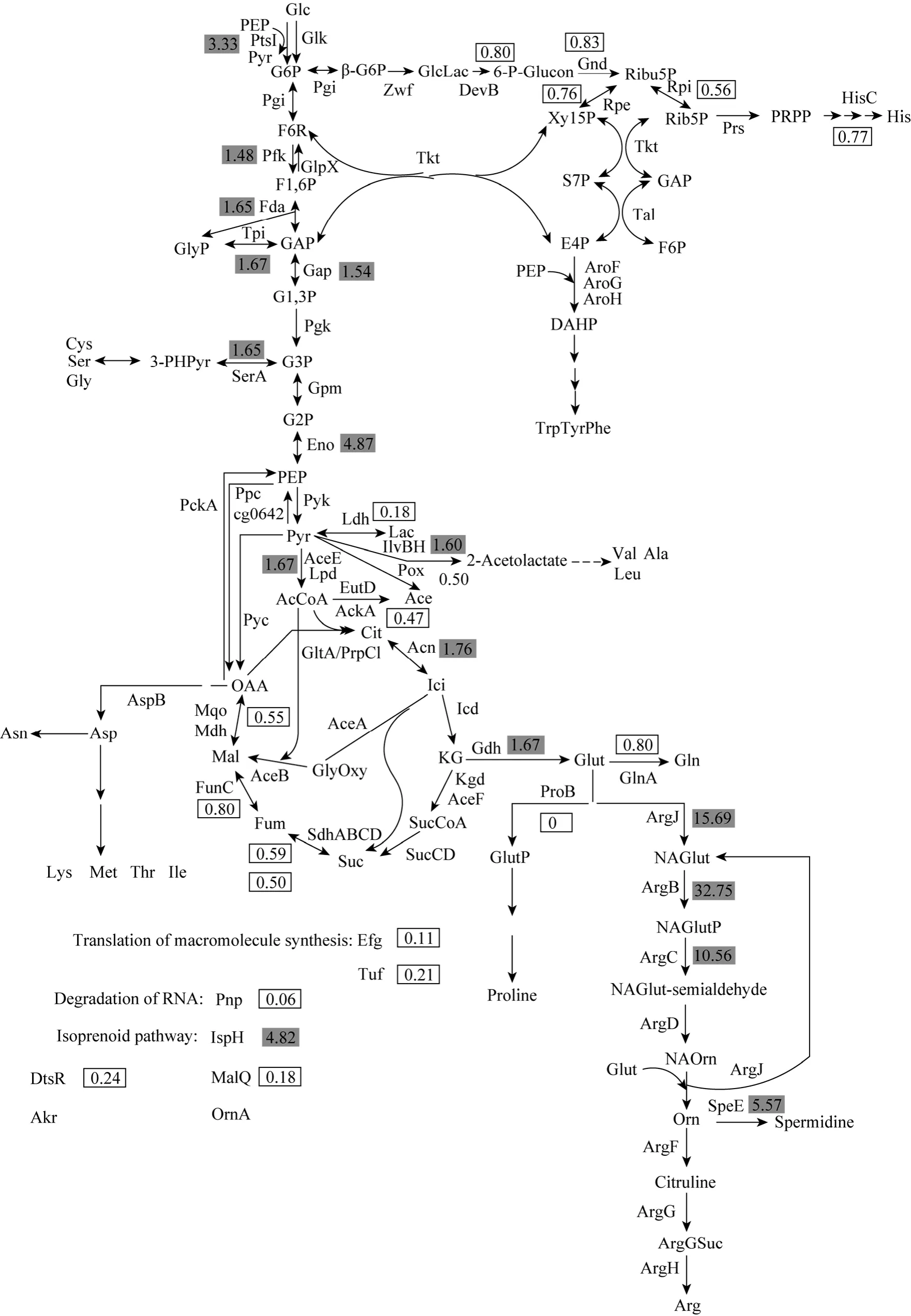
Figure 2 Metabolic map showing relative expression ratios of protein level at the exponential phases[The numbers are the ratio of the expression levels in C. glutamicum ATCC 13032 (ΔargF, ΔproB, Δkgd) versus C. glutamicum ATCC 13032. The shaded and boxed numbers indicate significantly up- and down-regulated proteins, respectively. Glc: glucose;G6P: D-glucose 6-phophate; F6P: D-fructose 6-phosphate; F1,6P: D-fructose 1,6-bisphosphate; GAP: D-glyceraldehyde
4 CONCLUSIONS
Metabolic engineering was used to improve L-ornithine production in C. glutamicum by disrupting genes to prevent the flux away from L-ornithine. It was also found that the inactivation of ODHC enhanced L-ornithine production, indicating that supply of endogenous glutamate by knockout of the kgd gene was beneficial for L-ornithine production. The comparative proteomic analysis between the wild-type and the engineered strain showed the cellular physiological changes and revealed the mechanism of L-ornithine overproduction of the engineered strain. Some probable strategies for further enhancing L-ornithine production were also proposed based on the results of comparative proteomic analysis. These strategies would be the bases for our further metabolic engineering studies of L-ornithine overproduction. The overexpression of the genes in the upstream pathway of glutamate to increase the availability of endogenous glutamate may further increase ornithine production in the engineered C. glutamicum and the ornithine synthesis enzymes (ArgCJBD) may not be the limiting enzymes in the engineered C. glutamicum. Blocking flux away from L-ornithine by disrupting genes (serA,ilvB and speE) may be another strategy for improving L-ornithine production.
1 Salvatore, F., Cimino, F., Maria, C., Cittadini, D., “Mechanism of the protection by L-ornithine-L-aspartate mixture and by L-arginine in ammonia intoxication”, Arch Biochem. Biophys., 107, 499-503 (1964).
2 Shi, H.P., Fishel, R.S., Efron, D.T., Williams, J.Z., Fishel, M.H.,Barbul, A., “Effect of supplemental ornithine on wound healing”, J.Surg. Res., 106, 299-302 (2002).
3 Zajac, A., Poprzecki, S., Zebrowska, A., Chalimoniuk, M., Langfort,J., “Arginine and ornithine supplementation increases growth hormone and insulin-like growth factor-1 serum levels after heavy-resistance exercise in strength-trained athletes”, J. Strength.Cond. Res., 24, 1082-1090 (2010).
4 Choi, D.K., Ryu, W.S., Choi, C.Y., Park, Y.H., “Production of L-ornithine by arginine auxotrophic mutants of Brevibacterium ketoglutamicum in dual substrate limited continuous culture”, J. Ferment. Bioeng., 81, 216-219 (1996).
5 Kinoshita, S., Nakayama, K., Udaka, S., “The fermentative production of L-ornithine”, J. Gen. Appl. Microbiol., 3, 276-277 (1957).
6 Zhang, J.F., Wang, J.B., Huang, J.M., Zhang, J., “Breeding of high-yield L-ornithine-producing strain by protoplast fusion”, Amino acids Biotic. Resour., 31, 53-57 (2009).
7 Sakanyan, V., Petrosyan, P., Lecocq, M., Boyen, A., Legrain, C.,Demarez, M., Hallet, J.N., Glansdorff, N., “Genes and enzymes of the acetyl cycle of arginine biosynthesis in Corynebacterium glutamicum: Enzyme evolution in the early steps of the arginine pathway”, Microbiology, 142, 99-108 (1996).
8 Lee, Y.J., Cho, J.Y., “Genetic manipulation of a primary metabolic pathway for L-ornithine production in Escherichia coli”, Biotechnol.Lett., 28, 1849-1856 (2006).
9 Hwang, J.H., Hwang, G.H., Cho, J.Y., “Effect of increased glutamate availability on L-ornithine production in Corynebacterium glutamicum”, J. Microbiol. Biotechnol., 18, 704-710 (2008).
10 Lee, S.Y., Cho, J.Y., Lee, H.J., Kim, Y.H., Min, J., “Enhancement of ornithine production in proline-supplemented Corynebacterium glutamicum by ornithine cyclodeaminase”, J. Microbiol. Biotechnol., 20,127-131 (2010).
11 Asakura, Y., Kimura, E., Usuda, Y., Kawahara, Y., Matsui, K.,Osumi, T., Nakamatsu, T., “Altered metabolic flux due to deletion of odhA causes L-glutamate overproduction in Corynebacterium glutamicum”, Appl. Environ. Microbiol., 73, 1308-1319 (2007).
12 Kim, J., Hirasawa, T., Sato, Y., Nagahisa, K., Furusawa, C., Shimizu,H., “Effect of odhA overexpression and odhA antisense RNA expression on Tween-40-triggered glutamate production by Corynebacterium glutamicum”, Appl. Microbiol. Biotechnol., 81, 1097-1106(2009).3-phosphate; GlyP: glycerone phosphate; G1,3P: 3-phospho-D-glyceroyl phosphate; G3P: 3-phospho-D-glycerate; G2P:2-phospho-D-glycerate; PEP: phosphoenolpyruvate; Pyr: pyruvate; Lac: lactate; Ace: acetate; AcCoA: Acetyl-CoA; Cit: citrate; Ici:isocitrate; KG: 2-oxoglutarate; SucCoA: succinyl-CoA; Suc: succinate; Fum: fumarate; Mal: malate; OAA: oxaloacetate; Glut: glutamate; GlutP: L-glutamyl 5-phosphate; NAGlut: N-acetyl-L-glutamate; NAGlutP: N-acetyl-L-glutamate 5-phosphate; NAGlutsemialdehyde: N-acetyl-L-glutamate 5-semialdehyde; NAOrn: N-acetylornithine; Orn: L-ornithine; ArgSuc: N-(L-argino)succinate;β-G6P: β-D-glucose 6-phophate; GlcLac: D-glucono-1,5-lactone 6-phosphate; 6-P-Glucon: 6-phospho-D-glucobate; Ribu5P:D-ribulose 5-phosphate; Xyl5P: D-xylulose 5-phosphate; Ribu5P: D-ribose 5-phosphate; S7P: sedoheptulose 7-phosphate; E4P:D-erythrose 4-phosphate; PRRP: 5-phospho-α-D-ribose 1-biphosphate; DAHP: 3-deoxy-D-arabino-hept-2-ulosonate 7-phosphate;3-PHPyr: 2-(α-hydroxyethyl)thiamine diphosphate; PtsI: phosphoenolpyruvate: sugar phosphotransferase system enzyme I; Glk:glucokinase; Pgi: glucose-6-phosphate isomerase; Pfk: fructose-bisphosphate aldolase; GlpX: fructose 1,6-bisphosphatase II; Fda:fructose-bisphosphate aldolase; Tpi: triosephosphate isomerase; Gap: glyceraldehyde-3-phosphate dehydrogenase A; Pgk:phosphoglycerate kinase; Gpm: phosphoglycerate mutase; Eno: enolase; Pyk: pyruvate kinase; AceE: pyruvate dehydrogenase; Lpd:dihydrolipoamide dehydrogenase; SerA: D-3-phosphoglycerate dehydrogenase; Ppc: phosphoenolpyruvate carboxylase; Pck: phosphoenolpyruvate carboxykinase; cg0642: phosphoenolpyruvate synthase; Pyc: pyruvate carboxylase; Ldh: D-lactate dehydrogenase;Pox: pyruvate dehydrogenase; EutD: phosphate acetyltransferase; AckA: acetate kinase; ilvBH: acetolactate synthase; GltA: citrate synthase II; PrpC1: citrate synthase; Acn: aconitate hydrase; Icd: isocitrate dehydrogenase; Kgd: α-ketoglutarate decarboxylase;AceF: dihydrolipoamide acetyltransferase; SucCD: succinyl-CoA synthetase, beta subunit; alpha subunit; SdhABCD: succinate dehydrogenase, flavoprotein subunit, FeS subunit, membrane subunit, hydrophobic subunit; FumC: fumarate hydratase; Mqo: malate:quinone oxidoreductase; Mdh: malate dehydrogenase; AspB: aspartate aminotransferase; AceA: isocitrate lyase; AceB: malatesynthase A; Gdh: glutamate dehydrogenase; GlnA: glutamine synthetase I; ProB: gamma-glutamyl kinase; ArgJ: N-acetylglutamate synthase;ArgB: acetylglutamate kinase; ArgC: N-acetyl-gamma-glutamyl-phosphate reductase; ArgD: acetylornithine aminotransferase; Spe:spermidine synthase; ArgF: ornithine carbamoyltransferase; ArgG: argininosuccinate synthase; ArgH: argininosuccinate lyase; Zwf:glucose-6-phosphate dehydrogenase; DevB: 6-phosphogluconolactonase; Gnd: gluconate-6-phosphate dehydrogenase; Rpe:D-ribulose-5-phosphate 3-epimerase; Rpi: ribose 5-phosphate isomerase B; Tkt: Transketolase; Tal: transaldolase; PrsA: phosphoribosylpyrophosphate synthase; AroFGH: 3-deoxy-D-arabino-heptulosonate-7-phosphate synthase; HisC: histidinol phosphate aminotransferase; Efg: elongation factor G; Tuf: elongation factor Tu; DtsR: acyl-CoA carboxylase; MalQ: 4-alpha-glucanotransferase;Akr:aldo/keto reductase; OrnA: oligoribonuclease; IspH: 4-hydroxy-3-methylbut-2-enyl diphosphate reductase]
13 Liu, J., Weng, Z., Wang, Y., Chao, H., Mao, Z., “Metabolic engineering based on systems biology for chemicals production”, Front.Biol. China, 4 (3), 260-265 (2009).
14 Zhang, Y., Liu, J.Z., Huang, J.S., Mao, Z.W., “Genome shuffling of Propionibacterium shermanii for improving vitamin B12 production and comparative proteome analysis”, J. Biotechnol., 148, 139-143(2010).
15 Lee, J., Lee, S.Y., “Proteome-based physiological analysis of the metabolically engineered succinic acid producer Mannheimia succiniciproducens LPK7”, Bioproc. Biosyst. Eng., 33, 97-107 (2010).
16 Kedar, P., Colah, R., Shimizu, K., “Proteomic investigation on the pyk-F gene knockout Escherichia coli for aromatic amino acid production”, Enzyme Microb. Technol., 41, 455-465 (2007).
17 Lee, J.H., Lee, D.E., Lee, B.U., Kim, H.S., “Global analyses of transcriptomes and proteomes of a parent strain and an L-threonineoverproducing mutant strain”, J. Bacteriol., 185, 5442-5451 (2003).
18 Park, J.H., Lee, S.Y., “Towards systems metabolic engineering of microorganisms for amino acid production”, Curr. Opin. Biotechnol.,19, 1-7 (2008).
19 Schäfer, A., Tauch, A., Jäger, W., Kalinowski, J., Thierbach, G.,Pühler, A., “Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum”, Gene, 145, 69-73 (1994).
20 Eikmanns, B.J., Thum-Schmitz, N., Eggeling, L., Ludtke, K.U.,Sahm, H., “Nucleotide sequence, expression, and transcription analysis of the Corynebacterium glutamicum gltA gene encoding citrate synthase”, Microbiology, 140, 1817-1828 (1994).
21 Van der Rest, M.E., Lange, C., Molenaar, D., “A heat shock following electroporation induces highly efficient transformation of Corynebacterium glutamicum with xenogeneic plasmid DNA”, Appl. Microbiol. Biotechnol., 52, 541-545 (1999).
22 Udaka, S., “Pathway-specific pattern of control of arginine biosynthesis in bacteria”, J. Bacteriol., 91, 617-621 (1966).
23 Haas, D., Leisinger, T., “N-acetylglutamate 5-phosphotransferase of Pseudomonas aeruginosa: purification and ligand-directed association-dissociation”, Eur. J. Biochem., 52, 365-375 (1975).
24 Bradford, M.M., “A rapid and sensitive method for the quantitation of microgram quantitites of protein utilizing the principle of protein-dye binding”, Anal. Biochem., 72, 248-254 (1976).
25 Schaffer, S., Weil, B., Nguyen, V.D., Dongmann, G., Gunther, K.,Nickolaus, M., Hermann, T., Bott, M., “A high-resolution reference map for cytoplasmic and membrane-associated proteins of Corynebacterium glutamicum”, Electrophoresis, 22, 4404-4422 (2001).
26 Schaffer, S., Bott, M., “Reference map for Corynebacterium glutamicum ATCC13032”, http://www.fz-juelich.de/ibt/cg-proteomic/(2007).
27 Chinard, F.P., “Photometric estimation of proline and ornithine”, J.Biol. Chem., 199, 91-95 (1952).
28 Kalinowski, J., Bathe, B., Bartels, D., Bischoff, N., Bott, M., Burkovski, A., Dusch, N., Eggeling, L., Eikmanns, B.J., Gaigalat, L.,Goesmann, A., Hartmann, M., Huthmacher, K., Kramer, R., Linke,B., McHardy, A.C., Meyer, F., Mockel, B., Pfefferle, W., Puhler, A.,Rey, D.A., Ruckert, C., Rupp, O., Sahm, H., Wendisch, V.F., Wiegrabe, I., Tauch, A., “The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of L-aspartate-derived amino acids and vitamins”, J. Biotechnol., 104,5-25 (2003).
29 Kawahara, Y., Takahashi-Fuke, K., Shimizu, E., Nakamatsu, T., Nakamori, S., “Relationship between glutamate production and the activity of 2-oxoglutarate dehydrogenase in Brevibacterium lactofermentum”, Biosci. Biotechnol. Biochem., 61, 1109-1112 (1997).
30 Shingu, H., Terui, G., “Studies on process of glutamic acid fermentation at the enzyme level. I. On the change of α-ketoglutaric acid dehydrogenase in the course of culture”, J. Ferment. Technol., 49,400-405 (1971).
31 Park, J.H., Lee, K.H., Kim, T.Y., Lee, S.Y., “Metabolic engineering of Escherichia coli for the production of L-valine based on transcriptome analysis and in silico gene knockout simulation”, Proc.Natl. Acad. Sci. USA, 104, 7797-7802 (2007).
32 Shirai, T., Fujimura, K., Furusawa, C., Nagahisa, K., Shioya, S.,Shimizu, H., “Study on roles of anaplerotic pathways in glutamate overproduction of Corynebacterium glutamicum by metabolic flux analysis”, Microb. Cell Fact., 6, 19 (2007).
33 Marx, A., Striegel, K., de Graaf, A.A., Sahm, H., Eggeling, L., “Response of the central metabolism of Corynebacterium glutamicum to different flux burdens”, Biotechnol. Bioeng., 56, 168-180 (1997).
34 Kimura, E., “L-glutamate production”, In: Handbook of Corynebacterium glutamicum, Eggeling, L., Bott, M., eds., CRS Press, Singapore, 439-464 (2005).
35 Bralley, P., Jones, G.H., “Overexpression of the polynucleotide phosphorylase gene (pnp) of Streptomyces antibioticus affects mRNA stability and poly(A) tail length but not ppGpp levels”, Microbiology, 149, 2173-2182 (2003).
36 Seibold, G.M., Wurst, M., Eikmanns, B.J., “Roles of maltodextrin and glycogen phosphorylases in maltose utilization and glycogen metabolism in Corynebacterium glutamicum”, Microbiology, 155,347-358 (2009).
37 Blombach, B., Seibold, G.M., “Carbohydrate metabolism in Corynebacterium glutamicum and applications for the metabolic engineering of L-lysine production strains”, Appl. Microbiol. Biotechnol.,86, 1313-1322 (2010).
 Chinese Journal of Chemical Engineering2012年4期
Chinese Journal of Chemical Engineering2012年4期
- Chinese Journal of Chemical Engineering的其它文章
- Phenol Oxidation by Combined Cavitation Water Jet and Hydrogen Peroxide*
- Venting Design for Di-tert-butyl Peroxide Runaway Reaction Based on Accelerating Rate Calorimeter Test
- Effect of Return Sludge Pre-concentration on Biological Phosphorus Removal in a Novel Oxidation Ditch*
- Separation of α-Tocopherol with a Two-Feed Simulated Moving Bed*
- Experimental and CFD Studies on the Performance of Microfiltration Enhanced by a Turbulence Promoter*
- Pervaporation of Aqueous Solution of Acetaldehyde Through ZSM-5 Filled PDMS Composite Membrane*