
大肠杆菌异源生产丁醇途径组装及启动子优化
2012-02-09 00:55:46唐玮李键陈军杨晟
生物工程学报 2012年11期
唐玮,李键,陈军,杨晟
中国科学院上海生命科学研究院植物生理生态研究所 中国科学院合成生物学重点实验室,上海 200032
通过优化启动子调节基因转录水平,疏通限速步骤,可达到提高目标产物产量的目的[1-2]。但是利用强启动子并不一定有效,因为一些目的基因的表达产物或中间代谢产物可能对菌体有毒性,从而抑制菌体生长[3-4],例如有研究表明在大肠杆菌中过表达编码磷酸烯醇式丙酮酸羧激酶的基因pck会给菌体生长带来负担[5];并且因为大多数的生物过程都是由多个基因相互影响的,很难通过调控一个基因得到最优化的途径。利用不同强度的启动子与不同基因的组合表达可以获得性状最优的组合[6]。大肠杆菌是常用的异源表达宿主[7],已有前人通过易错PCR获得了不同强度的启动子可用于途径优化,Alper和Braatsch等人构建了一系列不同强度的组成型启动子,其中 Alper系列包括强启动子 Alper PLTetO1和弱启动子Alper BB等,Alper PLTetO1的启动子强度是Alper BB的两倍[8],Braatsch系列包括强启动子Braatsch 20和弱启动子Braatsch 10等,Braatsch 20的启动子强度是Braatsch 10的两倍[9],可以利用这些不同强度的启动子与途径中不同的基因进行组合,通过检测产物获得最优化的菌株。
传统工业上利用丙酮丁醇梭菌生产丁醇[10-11],近年来通过大肠杆菌异源合成丁醇成为研究热点[12-13]。丁醇合成途径中相关的有编码硫解酶的thlA基因、bcs-operon以及醛/醇脱氢酶基因adhE1和adhE2。其中的bcs-operon由基因crt、bcd、etfAB、hbd组成,分别编码巴豆酸酶、丁酰辅酶 A脱氢酶,负责电子转移的黄素蛋白亚基,β-羟-丁酰辅酶A脱氢酶。有文献报道了ter基因编码的反式-2-烯酰辅酶 A还原酶 (trans-2-enoyl-CoA reductase),可以通过不可逆的反应为代谢流提供驱动力,替代丙酮丁醇梭菌来源bcd和etfAB可疏通大肠杆菌丁醇合成途径[14]。本研究用ter基因代替了bcd和etfAB构建了优化的bcs-operon (包括 crt、ter和 hbd基因,简称operon)。由于大肠杆菌内源的醇脱氢酶能够用于丁醇的合成[15],本研究将强启动子 Alper PLTetO1或弱启动子 Alper BB,强启动子Braatsch 20或弱启动子Braatsch 10分别与 thlA,operon进行组合,用DNA assembler方法[16]快速组装丁醇合成途径,获得最优的启动子组合的大肠杆菌丁醇生产菌株。这种启动子优化方法也可以在其他的途径优化中得到广泛的应用。
1 材料与方法
1.1 菌株和质粒
大肠杆菌Top10,酵母菌株BY4742和质粒pYES2均为本实验室保藏,pLY15质粒由本实验室构建和保藏,pMD18-T simple购自TaKaRa公司,启动子序列由GeneScript公司合成。
1.2 酶和试剂
限制性内切酶购自宝生物公司和Fermentas;质粒DNA小量试剂盒、PCR纯化试剂盒和DNA胶回收试剂盒购自Axygen,酵母质粒提取试剂盒购自索莱宝生物公司;琼脂购自上海捷倍思生物技术有限公司;其他试剂均为国产或进口分析纯。PCR反应使用的耐热DNA聚合酶为Taq (MBI Fermentas) 或者KOD (Toyobo)。
1.3 培养基
大肠杆菌培养使用 LB (Luria-Bertani) 培养基:胰化蛋白胨10 g、酵母提取物5 g、NaCl 10 g溶于1 L蒸馏水中,121 ℃高压蒸汽灭菌20 min。
酵母培养使用 YPD (Yeast extract peptone dextrose) 培养基:1%酵母提取物,2%胰化蛋白胨,2%葡萄糖,如果配成固体的再加2%的琼脂,121 ℃高压蒸汽灭菌20 min,其中葡萄糖需要单独灭菌。
酵母转化子筛选使用基本盐培养基(SC-ura3):1) 酵母基本氮源 (Yeast nitrogen base,YNB,无氨基酸或硫酸铵):0.34 g,硫酸铵:1 g,YNB+硫酸铵溶于50 mL ddH2O中,单独灭菌。2) 葡萄糖:4 g (溶于100 mL ddH2O中配成40%的溶液,单独灭菌。3) 琼脂4 g (溶于48 mL的ddH2O中单独灭菌)。4) 需要添加的氨基酸配成100×的母液,过滤除菌:12 mg组氨酸(His),18 mg赖氨酸 (Lys),60 mg亮氨酸 (Leu),溶于6 mL的ddH2O中,过滤除菌。在上述物品灭菌后,稍微冷却一下,将 YNB、硫酸铵和葡萄糖都加入液体琼脂中,再加入2 mL的过滤除菌的氨基酸混合液,倒板。
TB发酵培养基 (500 mL):1) 将下列组分溶解在300 mL ddH2O中:蛋白胨6 g,酵母提取物12 g,甘油2 mL。各组分溶解后高压灭菌,冷却到 60 ℃。2) 将 1.155 g的KH2PO4和 8.215 g K2HPO4溶在足量的 ddH2O 中,使终体积为100 mL。高压灭菌。3) 将10 g葡萄糖溶于100 mL ddH2O中,将3种溶液混合均匀。
1.4 方法
1.4.1 根据启动子和基因设计合适的RBS (核糖体结合位点) 序列
利 用 在 线 网 站 (https://salis.psu.edu/ software/forward) 设计最优的RBS序列。
1.4.2 设计同源臂引物
利用pLY15 (含有丁醇合成途径基因thlA以及operon) 质粒和人工合成的启动子为模板,设计同源臂,引物如表1所示。
1.4.3 制备启动子片段和丁醇合成途径的片段
用引物WA-BB-U和WA-BB-TLA-D扩增弱启动子片段Alper BB-RBS,引物SA-PLTetO1-U和SA-PLTetO1-TLA-D扩增强启动子片段Alper PLTetO1-RBS,引物WB-10-U和WB-10-CRT-D扩增弱启动子片段 Braatsch 10-RBS,引物SB20-U 和 SB20-CRT-D 扩增强启动子片段Braatsch 20-RBS。
用引物TLA-SA-U和TLA-SB-D扩增带有强启动子与强启动子组合的同源臂的 thlA片段(S-thlA-S),引物TLA-SA-U和TLA-WB-D扩增带有强启动子弱启动子组合的同源臂的 thlA片段 (S-thlA-W),引物TLA-WA-U和TLA-SB-D扩增带有弱启动子强启动子组合的同源臂的thlA片段 (W-thlA-S),引物 TLA-WA-U 和TLA-WB-D扩增带有弱启动子与弱启动子组合的同源臂的 thlA片段 (W-thlA-W),引物CRT-SB-U和HBD-pYES2-D 扩增带有强启动子同源臂的 operon 片段 (S-operon),引物CRT-WB10-U和HBD-pYES2-D扩增带弱启动子同源臂的operon片段 (W-operon),KOD酶扩增,PCR条件:95 ℃预变性5 min;94 ℃变性30 s,55 ℃退火45 s,68 ℃延2 min,35个循环;68 ℃延伸10 min,16 ℃保温10 min。

表1 本研究所用的引物Table 1 Primers used in the study
1.4.4 组建不同强度启动子的丁醇合成途径
用 EcoRⅠ酶切酵母-大肠杆菌穿梭载体pYES2 (氨苄抗性),回收线性化载体,回收Alper PLTetO1-RBS、AlperBB-RBS、Braatsch 20-RBS、Braatsch 10-RBS、S-thlA-S,S-thlA-W、W-thlA-S、W-thlA-W、S-operon、W-operon片段,进行不同强度的启动子和片段的组装,构建4种不同强度组合的质粒,如表2所示。
1.4.5 转化酵母BY4742
将构建4种质粒所需的回收片段进行混合,用核酸定量仪进行定量,载体片段约500 ng,其他片段为300 ng,用真空浓缩仪浓缩到4 μL,电转化酵母菌株BY4742[16],电击条件:电压1.5 kV,电容 25 μF,电阻 200 Ω,菌液涂布在SC-ura3的筛选板上,置于30 ℃培养箱中培养2~4 d会出现白色的明显菌落。
1.4.6 质粒PCR验证
挑取明显的酵母菌落,用酵母试剂盒提取酵母质粒,进行质粒PCR验证thlA以及operon片段。
1.4.7 酵母质粒转化大肠杆菌 Top10以及质粒的酶切验证
利用化学转化法转化构建的质粒到大肠杆菌 Top10中,用质粒提取试剂盒抽提质粒后用Hind Ⅲ进行酶切验证。
表2 不同强度的启动子与片段的组合Table 2 Combination of different promoters and fragments
1.4.8 丁醇含量测定
发酵方法:96孔板深孔发酵,培养基为TB发酵培养基,在微好氧、30 ℃的条件下发酵100 h。
丁醇含量测定条件:取上述样品清液0.2 mL与0.8 mL内标液混匀,以配置有Grace公司产品ECTM-WAX毛细管色谱柱的安捷伦公司 7890型气相色谱仪进行测定。进样量1 mL,分流比25∶1。分析条件为:柱温85 ℃,氮气34.5 kPa.,5.5 min后,升温至150 ℃;氮气206.85 kPa,保持3 min,氢气30 mL/min,空气400 mL/min,尾吹25 mL/min;进样区温度250 ℃,氢火焰检测区温度为300 ℃。
2 结果与分析
2.1 设计最优RBS序列
根据启动子和基因的组合设计最优的 RBS序列,结果如表3所示。
2.2 制备片段和线性化载体
扩增启动子 AlperPLTetO1-RBS、AlperBBRBS、Braatsch 20-RBS、Braatsch 10-RBS和丁醇合成途径相关基因片段 S-thlA-S、S-thlA-W、W-thlA-S、W-thlA-W、S-operon、W-operon,用EcoRⅠ酶切载体 pYES2,获得的片段纯化后电泳图谱如图1所示。
2.3 不同启动子强度的质粒构建
将纯化片段混合浓缩电转酵母BY4742,用DNA assembler方法组装不同强弱启动子组合的质粒pTY02、pTY03、pTY04和pTY05。提取酵母质粒进行质粒PCR,确定质粒的正确性。如图2和图3所示,证明thlA和operon片段同时存在于构建的质粒上。
表3 Alper系列和Braatsch系列的启动子序列以及最优RBS序列Table 3 Sequence of promoter and RBS

图1 利用设计的含有同源臂的引物PCR扩增启动子和丁醇合成途径基因Fig. 1 Amplification of promoter and butanol biosynthesis genes with the design of the containing homologous arm primer. 1: Alper PLTetO1-RBS; 2: AlperBB-RBS; 3: S-thlA-S; 4: S-thlA-W; 5: W-thlA-S; 6: W-thlA-W; 7: Braatsch 20-RBS; 8: Braatsch 10-RBS; 9: S-operon; 10: W-operon; 11: linear vector pYES2; M: DNA marker.
2.4 反转大肠杆菌后质粒Hind Ⅲ酶切验证
将酵母质粒反转大肠杆菌Top10后,抽提大肠杆菌质粒用Hind Ⅲ进行酶切验证。结果见图4,证明质粒构建正确。
2.5 发酵丁醇产量测定
挑取验证正确的大肠杆菌,利用 96孔板发酵100 h,用气相色谱测定丁醇产浓度,结果如图5所示,证明pTY03组合的质粒产丁醇浓度最高,达到28 mg/L,是其他启动子组合的3~5倍。
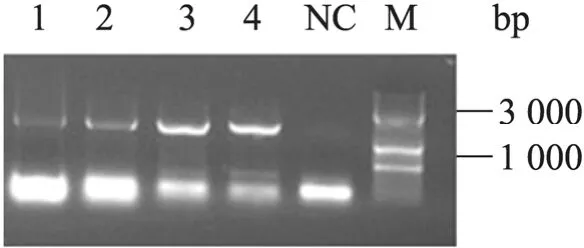
图2 质粒PCR验证组装的4种质粒的thlA片段Fig. 2 Identification of thlA fragment by PCR. 1: pTY02; 2: pTY03; 3: pTY04; 4: pTY05; NC: negative control; M: DNA marker.
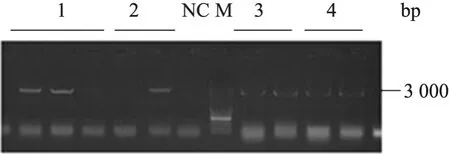
图3 质粒PCR验证组装的4种质粒的operon片段Fig. 3 Identification of operon fragment by PCR. 1: pTY02; 2: pTY03; 3: pTY04; 4: pTY05; NC: negative control; M: DNA marker.
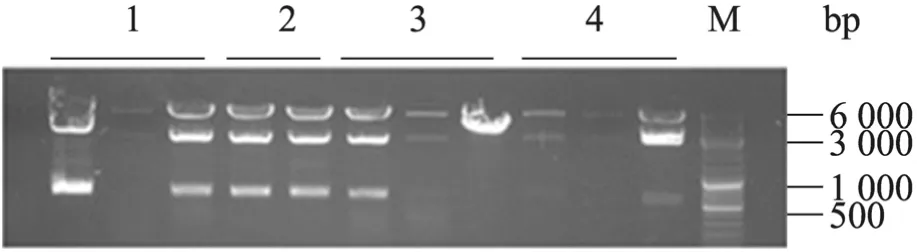
图4 Hind Ⅲ酶切验证丁醇途径组装质粒Fig. 4 Identification of butanol way assembly plasmid by enzyme digestion.

图5 含有 4种组合的质粒的大肠杆菌 Top10发酵100 h后的丁醇情况Fig. 5 Four different combination plasmids fermentation results by Escherichia coli Top 100 after 100 h. NC: negative control.
3 讨论
生物代谢途径很少由一个基因控制,大多都是通过多个基因的相互作用[17],所以代谢工程改造需要平衡代谢流以获得最优的效果[18-19]。通过强启动子提高转录水平不一定能获得最优的目标产物的产量,甚至会抑制宿主的生长。而通过不同强度启动子的精细调控可以更好地平衡多个基因的相互作用,获得最优的结果。本研究利用DNA assembler方法快速组装了不同强度启动子组合的丁醇合成途径。DNA assembler是一种利用酵母的高同源重组效率[20]从而把目的片段一次性在酵母体内进行组装的方法,耗时短,效率高,避免了传统的酶连技术所需的酶切连接等步骤,同时解除了酶切位点对操作的限制[21-22],更加符合合成生物学日益发展的需要[23]。但是进行组装的片段不能出现较长的相同序列,以免发生重组,引起片段缺失。本研究在梭菌天然生产丁醇的途径基础上进行改造,利用大肠杆菌启动子库,优化RBS序列,将thlA基因以及构建的优化的 operon基因与不同强度的启动子进行组合,根据最后的发酵结果可以看出 pTY03质粒丁醇含量最高,达到28 mg/L,与其他的启动子组合相比丁醇产量提高了3~5倍。这可能是因为thlA是整个途径中的第一步,thlA的强转录保证了前体的供应充足,而丁醇的产生会给宿主带来负担,甚至影响宿主的生长,用弱启动子转录后续丁醇合成操纵子相对更好地平衡了代谢流,达到最好的效果。另外,由于同一个operon上的3个基因表达强度不一定一致,我们可以通过将operon上的基因进行重排,或者用单顺反子的形式进行基因的进一步细化表达。除启动子优化外,还有一些常用于提高异源表达产物量的方法[24]:1) 敲除副产物途径和竞争途径的基因,将代谢流尽可能引向目标产物[25];2) 考虑异源表达的酶之间的相互作用,通过酶的天然选择来达到提高目标产物的目的[26];3) 优化表达,对表达基因进行密码子优化,修饰SD序列和RBS位点[27];4) 蛋白质工程方面的改造[28-29],包括有理设计[30]和无理设计[31];5) 发酵工程方面改造,改变发酵条件和底物等,提高产物产量等。如进一步优化大肠杆菌异源生产丁醇途径,则可考虑通过启动子优化与酶的天然组合、支路途径敲除、过表达、发酵条件优化[12]等代谢工程手段结合进一步提高丁醇产量。
REFERENCES
[1] Nevoigt E, Kohnke J, Fischer CR, et al. Engineering of promoter replacement cassettes for fine-tuning of gene expression in Saccharomyces cerevisiae. Appl Environ Microbiol, 2006, 72(8): 5266−5273.
[2] Jensen PR, Hammer K. Artificial promoters for metabolic optimization. Biotechnol Bioeng, 1998, 58(2/3): 191−195.
[3] Jin YS, Ni HY, Laplaza JM, et al. Optimal growth and ethanol production from xylose by recombinant Saccharomyces cerevisiae require moderate D-xylulokinase activity. Appl Environ Microbiol, 2003, 69(1): 495−503.
[4] Gorgens JF, van Zyl WH, Knoetze JH, et al. The metabolic burden of the PGK1 and ADH2 promoter systems for heterologous xylanase production by Saccharomyces cerevisiae in defined medium. Biotechnol Bioeng, 2001, 73(3): 238−245.
[5] Chao YP, Patnaik R, Roof WD, et al. Control of gluconeogenic growth by pps and pck in Escherichia coli. J Bacteriol, 1993, 175(21): 6939−6944.
[6] Lu C, Jeffries T. Shuffling of promoters for multiple genes to optimize xylose fermentation in an engineered Saccharomyces cerevisiae strain. Appl Environ Microbiol, 2007, 73(19): 6072−6077.
[7] Clomburg JM, Gonzalez R. Biofuel production in Escherichia coli: the role of metabolic engineering and synthetic biology. Appl Microbiol Biotechnol, 2010, 86(2): 419−434.
[8] Alper H, Fischer C, Nevoigt E, et al. Tuning genetic control through promoter engineering. Proc Natl Acad Sci USA, 2005, 102(36): 12678−12683.
[9] Braatsch S, Helmark S, Kranz H, et al. Escherichia coli strains with promoter libraries constructed by Red/ET recombination pave the way for transcriptional fine-tuning. Biotechniques, 2008, 45(3): 335−337.
[10] Lee SY, Park JH, Jang SH, et al. Fermentative butanol production by Clostridia. Biotechnol Bioeng, 2008, 101(2): 209−228.
[11] Nolling J, Breton G, Omelchenko MV, et al. Genome sequence and comparative analysis of the solvent-producing bacterium Clostridium acetobutylicum. J Bacteriol, 2001, 183(16): 4823−4838.
[12] Atsumi S, Cann AF, Connor MR, et al. Metabolic engineering of Escherichia coli for 1-butanol production. Metab Eng, 2008, 10(6): 305−311.
[13] Inui M, Suda M, Kimura S, et al. Expression of Clostridium acetobutylicum butanol synthetic genes in Escherichia coli. Appl Microbiol Biotechnol, 2008, 77(6): 1305−1316.
[14] Shen CR, Lan EI, Dekishima Y, et al. Driving forces enable high-titer anaerobic 1-butanol synthesis in Escherichia coli. Appl Environ Microb, 2011, 77(9): 2905−2915.
[15] Berezina OV, Zakharova NV, Brandt A, et al. Reconstructing the clostridial n-butanol metabolic pathway in Lactobacillus brevis. Appl Microbiol Biotechnol, 2010, 87(2): 635−646.
[16] Shao Z, Zhao H. DNA assembler, an in vivo genetic method for rapid construction of biochemical pathways. Nucleic Acids Res, 2009, 37(2): e16.
[17] Schaaff I, Heinisch J, Zimmermann FK. Overproduction of glycolytic-enzymes in yeast. Yeast, 1989, 5(4): 285−290.
[18] Kauffman KJ, Prakash P, Edwards JS. Advances in flux balance analysis. Curr Opin Biotech, 2003, 14(5): 491−496.
[19] Nielsen J. It is all about metabolic fluxes. J Bacteriol, 2003, 185(24): 7031−7035.
[20] Iizasa E, Nagano Y. Highly efficient yeast-based in vivo DNA cloning of multiple DNA fragments and the simultaneous construction of yeast/Escherichia coli shuttle vectors. Biotechniques, 2006, 40(1): 79−83.
[21] DeJong JM, Liu YL, Bollon AP, et al. Genetic engineering of Taxol biosynthetic genes in Saccharomyces cerevisiae. Biotechnol Bioeng, 2006, 93(2): 212−224.
[22] Yan YJ, Kohli A, Koffas MAG. Biosynthesis of natural flavanones in Saccharomyces cerevisiae. Appl Environ Microb, 2005, 71(9): 5610−5613.
[23] Keasling JD. Synthetic biology for synthetic chemistry. Acs Chem Biol, 2008, 3(1): 64−76.
[24] Keasling JD, Chou H. Metabolic engineering delivers next-generation biofuels. Nat Biotechnol, 2008, 26(3): 298−299.
[25] Fell DA, Thomas S. Physiological control of metabolic flux-the requirement for multisite modulation. Biochem J, 1995, 311(Pt1): 35−39.
[26] Steen EJ, Chan R, Prasad N, et al. Metabolic engineering of Saccharomyces cerevisiae for the production of n-butanol. Microbial Cell Factories, 2008, 7: 36.
[27] Park YS, Seo SW, Hwang S, et al. Design of 5'-untranslated region variants for tunable expression in Escherichia coli. Biochem Biophys Res Commun, 2007, 356(1): 136−141.
[28] Leisola M, Turunen O. Protein engineering: opportunities and challenges. Appl Microbiol Biotechnol, 2007, 75(6): 1225−1232.
[29] Chiang SJ. Strain improvement for fermentation and biocatalysis processes by genetic engineering technology. J Ind Microbiol Biot, 2004, 31(3): 99−108.
[30] Machielsen R, Looger LL, Raedts J, et al. Cofactor engineering of Lactobacillus brevis alcohol dehydrogenase by computational design. Eng Life Sci, 2009, 9(1): 38−44.
[31] Petrounia IP, Arnold FH. Designed evolution of enzymatic properties. Curr Opin Biotechnol, 2000, 11(4): 325−330.
猜你喜欢
——紫 苏
河南农业(2024年1期)2024-01-19 01:56:54
华人时刊(2023年1期)2023-03-14 06:43:36
汉字汉语研究(2021年2期)2021-08-30 08:58:46
中国调味品(2017年2期)2017-03-20 16:18:25
创新作文(小学版)(2016年16期)2016-11-11 05:47:54
现代检验医学杂志(2016年5期)2016-08-20 03:17:04
河北书画研究(2016年3期)2016-04-28 08:55:35
中国科技信息(2015年2期)2015-11-16 08:18:32
结核与肺部疾病杂志(2015年2期)2015-07-18 11:00:27
河南科技(2014年12期)2014-02-27 14:10:24
