
Three-dimensional structures of virulence proteins of Legionella establish targets for new antibacterials
2012-01-23 01:12GuidoHansenRolfHilgenfeld
微生物与感染 2012年1期
Guido Hansen, Rolf Hilgenfeld
1. Institute of Biochemistry, Center for Structural and Cell Biology in Medicine, University of Lübeck, Lübeck 23538, Germany; 2. German Centre for Infection Research (DZIF), University of Lübeck, Lübeck 23538, Germany; 3. Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China; 4. Laboratory for Structural Biology of Infection and Inflammation, c/o DESY, Hamburg 22603, Germany
1 Introduction
In 1977,Legionellapneumophila(L.pneumophila) was identified as the causative agent of an atypical and severe form of pneumonia designated Legionnaires’ disease[1]. The bacteria have since been recognized as an increasingly important pathogen in health-care-, community-, and domestically-acquired pneumonia.Legionellaspp. are Gram-negativeγ-proteobacteria that parasitize protozoan host cells such asAcanthamoeba,Hartmannella, andTetrahymenain their natural fresh-water habitat. In addition, manyLegionellaspecies are able to efficiently multiply in artificial warm-water systems such as spa pools, air-conditioners, indoor fountains, and cooling towers. As many of these devices produce aerosols that are potentially inhaled,Legionellacan enter human hosts and cause infections after uptake into alveolar macrophages[2]. However, only pathogenicLegionellastrains are able to avoid phagosome-lysosome fusion and replicate inside human host cells. The interaction of virulentLegionellawith phagocytic host cells involves several steps: (1) adhesion to the host-cell surface; (2) uptake; (3) escape from the innate immune response; (4) establishment of a replicative vacuole; and (5) intracellular multiplication and egress from the host cells[3]. However, the underlying virulence mechanisms are complex and far from being fully understood.
X-ray crystallography has proved a valuable tool to reveal the molecular basis of virulence in a number of important pathogens. To date, structures of 46Legionellaproteins are available in the Protein Data Bank, yet, only 10 of those represent confirmed virulence factors. Thus, structural information ofLegionellaproteins important for pathogenicity is still scarce, despite considerable progress during the last decade. Here, we review two structures ofLegionellavirulence proteins, FeoB and DegQ, determined very recently. In addition, we discuss the best characterized virulence protein ofLegionella, the macrophage infectivity potentiator (Mip) protein, which was the firstLegionellaprotein with known three-dimensional structure. In contrast to Mip, a confirmed drug target, the structures of FeoB and DegQ present new potential targets with unique possibilities for the development of effective antibacterials.
2 Mip, FeoB and DegQ
2.1 Mip
Mip is a virulence protein that has been found in several intracellular pathogens such asL.pneumophila,Chlamydiaspp.,Neisseriagonorrhoeae,Trypanosomacruzi, andBurkholderiapseudomallei[4-8]. Its key feature is an intrinsic peptidyl-prolylcis/transisomerase (PPIase; EC 5.2.1.8) activity which is conferred by a FK506-binding protein (FKBP) domain. This activity is efficiently inhibited by the immunosuppressive drugs FK506 (tacrolimus) and rapamycin (sirolimus)[9]. The C-terminal FKBP domain of Mip fromL.pneumophila(LpMip) shares ~35% amino acid sequence identity with human FKBP12 and is required for virulence[10,11].L.pneumophilamutants lacking Mip cannot efficiently infect human macrophages[4]or mononuclear phagocytes[12], and show suboptimal growth in the fresh-water host organismsHartmannellaandTerahymena[13]. In a guinea pig model system for Legionnaires’ disease, it has been shown that Mip contributes to successful dissemination ofL.pneumophilathroughout the lung, most likely by interacting with collagen IV[11]. Involvement in host-cell infection has also been reported for Mip proteins from other pathogens[6,7,14].
LpMip was the first Mip protein to be structurally characterized. At a resolution of 2.4 Å, the X-ray crystal structure revealed thatLpMip forms a non-globular V-shaped homodimer which is stabilized exclusively by contacts between the N-terminal domains of twoLpMip molecules[15](Fig.1A). Dimer formation depends on the interaction of helices α1 and α2 of oneLpMip molecule with the equivalent helices of the other, together forming a shared antiparallel four-helix bundle. Most of the interactions between the helices are of hydrophobic nature and include a feature that we called a “methionine zipper”. A linker helix of 65 Å length connects the N-terminal domain with the C-terminal FKBP domain. PPIase activity and dimerization ofLpMip is essential for efficient multiplication inAcanthamoebaand for full virulence in the guinea pig model system[16]. Very similar results have been reported for the Mip homologue FKBP22 fromShewanellasp.[17]. In both systems, engineered monomeric protein variants were used to show that binding affinity and PPIase activity on protein substrates is strongly dependent on dimerization[16,17]. However, the molecular basis for these findings is not fully understood. The close proximity of two ligand-binding sites located in the FKBP domains might offer an advantage in the competition with host FKPBs for substrates. It is also possible that dimeric V-shaped FKPB proteins likeLpMip might ‘embrace’ substrate proteins with their long linker helices to bring the catalytic FKBP domains into position. Interestingly, the Mip homologue inTrypanosomacruzi(TcMip) does not seem to depend on dimerization.Trypanosomacruzicauses Chagas’ disease (American trypanosomiasis), which, according to the World Health Organization (WHO), afflicts 10 million people in Central and South America. The crystal structure ofTcMip revealed a common FKBP core shared withLpMip[18]. However, significant structural variations are present in regions N- and C-terminal of this core region. Most importantly,TcMip lacks the two helices α1 and α2 necessary for the formation of the four-helix bundle that is responsible for the formation of theLpMip dimer[15,18]. As a result,TcMip is monomeric. UnlikeLegionella,Trypanosomasecrete Mip into the medium prior to uptake into host cells, in order to increase infectivity[7]. Thus, the observed structural differences betweenLpMip andTcMip might reflect distinct functions during the life cycle of both pathogens. However, in both species, the precise molecular mechanism of action responsible for the Mip-dependent increase of virulence remains elusive and has to be addressed in future experiments.
X-ray and nuclear magnetic resonance (NMR) structures of FK506 and rapamycin in complex with the FKBP domain ofLpMip allowed the identification and detailed characterization of the ligand-binding site[15,19]. Both ligands bind to a hydrophobic pocket between the central β-sheet and helix α4 of the FKBP domain. This pocket accommodates the pipecoline ring of rapamycin (Fig.1B) and FK506[15,19]. Other groups of the ligands involved in interactions with the protein are the ester linkage, the dicarbonyl group, and the pyranosyl ring (Fig.1B). As most residues of the ligand-binding site are well conserved in Mip proteins from different species, it can be assumed that, in general, a given drug molecule exhibits a single, well-defined binding mode shared by most if not by all Mip proteins. It is therefore likely that new antibacterials targeting Mip will be useful for the therapy of a diverse set of bacterial infections, making the development of such drug molecules even more rewarding. As FK506 and rapamycin have immunosuppressive properties, these drugs themselves may not be suitable for treating bacterial infections.

A: LpMip dimer with transparent surface displaying the N-terminal dimerization domain, the long linker helix α3 and the C-terminal FBKP domain; PDB code: 1FD9[15]. B: LpMip FKBP domain (surface representation; blue) in complex with rapamycin (stick representation; carbon: yellow; oxygen: red; nitrogen: blue), parts of rapamycin in contact with LpMip indicated; PDB code: 2VCD[19].
However, structural information derived from the available ligand complexes is vital for the rational design of inhibitory molecules that lack unfavorable effects on the immune system. On the basis of structural and biochemical data, a first step towards the development of selective low-molecular-weight Mip inhibitors has recently been reported[20]. In this approach, a series of compounds sharing a common pipercoline moiety as an anchoring group were evaluated with respect to their ability to inhibit the PPIase activity ofLpMip. As these compounds lack the macrocyclic portion of rapamycin, it is unlikely that they will exhibit immunosuppressive effects. A promising candidate for further lead development was identified[20]and awaits further characterization in cell-based and guinea pig model systems.
2.2 Ferrous iron transporter (FeoB)
Availability of sufficient amounts of iron is critical for optimal growth of many bacterial species. In fact, depletion of free iron is an elegant way of eukaryotic cells to prevent the replication of invading pathogens. To counteract this strategy, many pathogenic bacteria have evolved systems to efficiently scavenge ferric (Fe3+) as well as ferrous (Fe2+) iron. InLegionella, the secreted low-molecular-weight siderophore, legiobactin, chelates Fe3+, following re-uptake of the complexed metal by a specific active transport mechanism[21]. Ferrous iron (Fe2+) is critical forL.pneumophilagrowth under low-oxygen conditions in host cells and in the mammalian lung[22]. The transmembrane protein FeoB is responsible for Fe2+uptake and has been shown to contribute to virulence in a number of pathogenic bacteria[22,23]. It consists of an intracellular N-terminal region of ~270 amino acid residues (NFeoB) and a C-terminal membrane-embedded domain of ~500 residues (CFeoB). Biochemical studies suggested that NFeoB harbors a GTP-binding/GTPase domain (G domain) and an additional domain which functions as guanine nucleotide dissociation inhibitor (GDI)[24].
Recently, we have determined the structure of NFeoB fromL.pneumophila(NFeoBLp) to a resolution of 2.5 Å[25]. NFeoB is a monomeric GTPase with characteristic G domain fold and an additional intramolecular GDI domain (Fig.2A). Interestingly and unusually for GTPases, the G5 motif, which is typically involved in the recognition of the guanine base in the nucleotide-binding site, adopts a closed conformation even in ligand-free state. Furthermore, the structure suggests how conformational changes upon nucleotide binding might affect the associated GDI and transmembrane regions to facilitate the regulated uptake of Fe2+.
Before 2009, no structural information on NFeoB from any species was available. However, the simultaneous release of structures of NFeoB fromEscherichiacoli(E.coli) (PDB codes: 3I8S, 3I8X, and 3I92) by our group and of the FeoB G domain fromMethanococcusjannaschii(PDB codes: 2WJG, 2WJH, 2WJI, and 2WJJ) by Köster,etal.[26]was closely followed by a remarkable avalanche of structures in the field. In fact, 14 new coordinate sets comprising NFeoB from five additional bacterial species (Thermotogamaritima[27];L.pneumophila[25];Streptococcusthermophila[28];Pyrococcusfuriosus[29]; andKlebsiellapneumoniae[29];E.coli[30]) have been published subsequently (Tab.1). Moreover, in many cases NFeoB proteins have been crystallized in ligand-free as well as in nucleotide-bound forms, facilitating the detailed analysis of the effect of GTP binding and hydrolysis. Therefore, today, NFeoB can be regarded as a structurally very well characterized system.
The overall fold of NFeoB is identical in all species (root mean square deviation typically <1 Å). However, as the structures show important differences in functionally relevant elements and in their oligomerization mode, conflicting mechanistic models for NFeoB have been proposed. In the following, the most striking deviations are presented and briefly discussed:

Fig.1 Structural data on FeoB proteins
* published coordinate sets and structure factors only. Work from our laboratory.
(1) Regions typically involved in nucleotide recognition in eukaryotic G proteins lack defined conformational changes in different ligation states of most NFeoBs. For instance, the switch I element, which is responsible for the interaction with theγ-phosphate of GTP and a bound Mg2+ion in prototypical GTPases, is either flexible or adopts a conformation usually found in the GDP-bound or ligand-free state. This interesting feature offers an explanation for the relatively weak nucleotide-binding affinity of NFeoB when compared to eukaryotic GTPases. Although there is some evidence that switch I might contain a Fe2+or potassium-binding site[28,29], its function is unclear. Surprisingly, in NFeoB fromStreptococcusthermophilain complex with mant-GMPPNP, the typical conformation with switch I contacting the nucleotide was observed (Fig.2B). Therefore, it cannot be fully excluded that switch I in NFeoB functions as nucleotide sensor and that the unusual conformation of switch I in the GTP-bound state found in most NFeoB structures represents an artifact caused by crystal contacts or the absence of an essential co-factor (such as potassium or Fe2+) during crystallization. (2) The molecular assembly found in crystals of ligand-free and nucleotide-bound forms of NFeoB fromE.coligave rise to a compelling mechanistic model. According to this model, three NFeoB molecules oligomerize to allow Fe2+ions to access a central channel of about 20 Å length, that facilitates regulated iron uptake. In ligand-free and GDP-bound forms of NFeoB, this channel is blocked, while a narrow opening was observed in the mant-GTP-bound structure. A similar arrangement of molecules has also been found in crystals of ligand-free and GTP-analog-bound NFeoB ofKlebsiellapneumoniae[29], supporting this so-called trimer-Fe2+-gating model. However, the remaining 14 NFeoB structures including NFeoBLpdo not display such trimeric assemblies, but form monomers or different dimeric species. The strong conservation of structure within the FeoB family suggests that iron uptake follows a common mechanism and hence for the trimer-Fe2+-gating model to be correct, and the trimer should be the dominating oligomeric species in most if not all crystal forms of NFeoB. This is obviously not the case. Notably, even for NFeoB proteins that form trimers in the crystal lattice, such trimers have never been confirmed to exist in solution. These results therefore raise questions about the validity of trimer-Fe2+-gating in general or its applicability to all NFeoB proteins.

A: Overall structure of NFeoBLp as cartoon representation including G domain (blue) with switches I (green) and II (orange), inter-domain linker (gray) and GDI domain (red); PDB code: 3IBY[25]. B: Cartoon representation of NFeoBSt (colors as in A) in complex with GMPPNP (sticks; carbon: yellow; oxygen: red; nitrogen: blue; phosphorus: orange) and Mg2+ (sphere; purple) showing a closed conformation with switch I contacting the nucleotide; PDB code: 3LX5[28].
2.3 Protein quality control protein (DegQ)
Besides overcoming the shortage of iron and other essential nutrients, intracellularLegionellahave to evade defense mechanisms of the host cells aiming to actively destroy invading pathogens. In hostile environments as encountered in phagosomes of professional macrophages,Legionellaneeds to prevent the excessive accumulation of misfolded proteins in the periplasm. In many prokaryotes, members of the HtrA family of proteins deal with this problem, promoting correct folding or efficient removal of misfolded or damaged periplasmatic proteins[31]. Accordingly, HtrA proteins have been identified as virulence proteins affecting intracellular survival of many pathogenic bacteria[32]. DegQ is an HtrA-family member with dual functions, combining chaperone and protease activities to facilitate refolding or degradation of misfolded proteins, respectively. DegQ is related to DegP, which represents a second HtrA protein with partially overlapping functions[33]. Both proteins share a common domain organization with an N-terminal trypsin-like protease domain preceding two PDZ domains (PDZ1 and PDZ2). Whereas inE.coliDegP and DegQ are present, many prokaryotes includingLegionellaspp. lack a DegP homologue, stressing the importance of DegQ for protein homeostasis in the periplasm.
Very recently, we reported the X-ray crystal structure of DegQ fromLegionellafallonii(DegQLf) at a resolution of 2.15 Å[34]. Interestingly, DegQLfforms large oligomers consisting of 12 protein molecules in solution as well as in the crystal lattice (Fig.3A). The DegQLf12-mer displays tetrahedral symmetry and is composed of 4 tightly interlocked homotrimers as basic building blocks. Whereas these homotrimers are stabilized by interactions between three protease domains, and formation of the 12-mer depends on contacts between PDZ domains of neighboring 3-mers. The overall organization of the particle resembles a hollow sphere with a protein shell enclosing a large internal cavity of ~70 Å diameter. All 12 protease active sites line the inner wall of the particle and are therefore not directly accessible from the outside. However, six lateral pores (~14 Å x ~28 Å) located in the protein shell connect the internal cavity with bulk solvent. To probe the functional properties of the DegQ 12-mer, we designed truncated protein variants that lack the C-terminal PDZ2 domain (DegQLfΔPDZ2) or both PDZ domains (DegQLfΔPDZ1&2). As expected from the crystal structure, both variants were unable to form 12-mers and were proteolytically inactive. However, chaperone activity was not affected. Thus, in DegQLf, the PDZ domains are necessary for 12-mer formation which in turn is essential for protease but not for chaperone activity. Results from another DegQLfvariant lacking specific residues of PDZ2 stabilizing the 12-mer confirmed these findings[34].
The structure of DegQLfshows that many aspects of architecture are shared between HtrA family members but also reveals that regulation of protease activity is fundamentally different in DegQ and DegP. In the well-characterized DegP system, 6-mers[35](Fig.3B) represent important protease-resting states, preventing deleterious proteolytic activity. The presence of unfolded proteins triggers the disassembly of 6-mers into two 3-mers and subsequent reassembly into active 12-mers or 24-mers[36-38](Fig.3C), dependent on the size and the concentration of the substrates. These larger oligomeric forms are responsible for the degradation of defective proteins under stress conditions[36,38]. Although, as in DegP, 12-mers are the protease-active form of DegQLf, we could not find any experimental evidence for DegQLf6-mers to exist. Instead, in the absence of unfolded proteins, DegQLf12-mers, 3-mers and a smaller fraction of monomers were observed[34]. The protease active sites in DegQLf3-mers are accessible, thus potentially harmful proteolytic activity needs be controlled in a different manner as in DegP 6-mers. To understand why 3-meric forms of DegQLfare inactive, we determined the structure of DegQLfΔPDZ2. This structure revealed an intrinsic flexibility of the remaining PDZ1 domain, allowing a rotation of approximately 180° with respect to the protease domain. This rotation places the protease active site and a regulatory substrate-binding site located in the PDZ1 domain on opposite faces of the 3-mer, effectively shutting down proteolytic activity. DegQLfshares a second regulatory mechanism with other HrtA family members. The protease active site in substrate-free HtrA proteins predominantly adopts a distinct OFF conformation, with the substrate binding cleft blocked by loop L1. A productive ON conformation can be assumed only after binding of an unfolded substrate to the regulatory site of a neighboring PDZ1 domain. As a result, an intrinsic activation cascade is triggered along loops L3, LD, and L1/L2, which are located in between PDZ1 and protease active site. Very similar molecular-switch mechanisms have been found in DegS[39], DegP[40], and Deg1 ofArabidopsisthaliana[41].
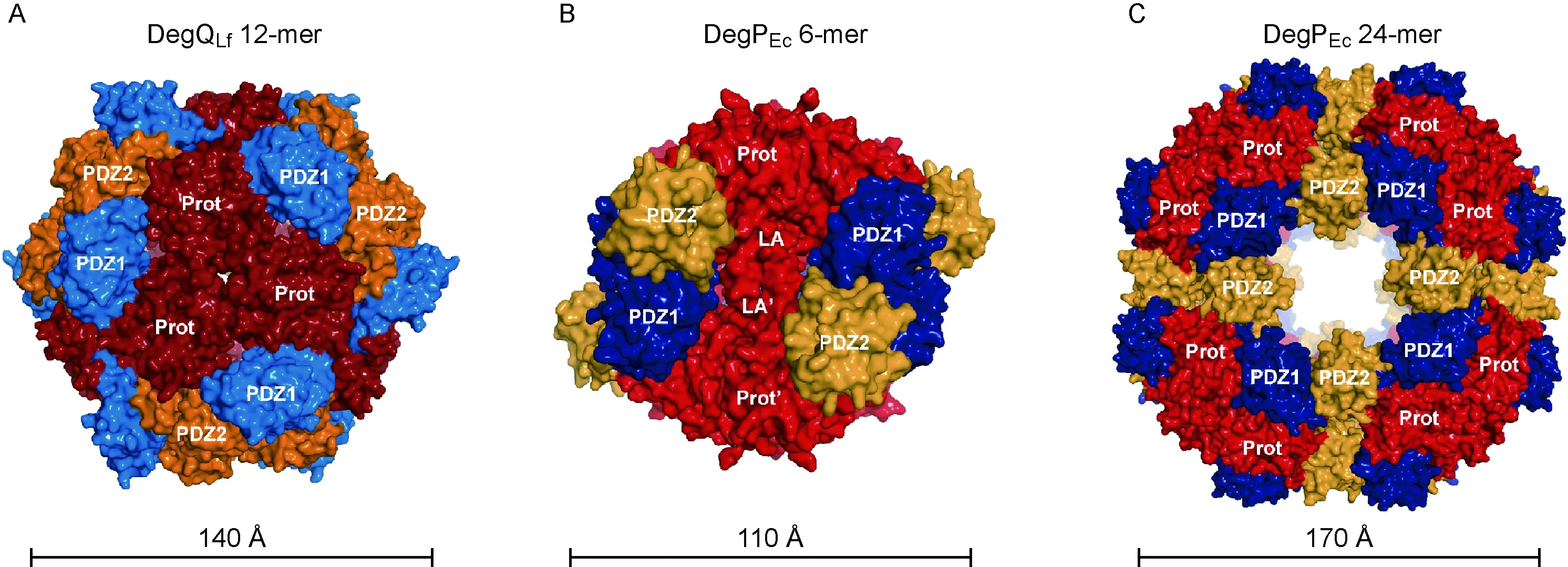
A: DegQLf 12-mer. View along the threefold axis at the protease interface with protease domain (Prot, dark red), PDZ1 domain (blue), and PDZ2 domain (orange); PDB code: 3PV2[34]. B: DegPEc 6-mer, interacting LA loops of opposing 3-mer that stabilize the 6-mer indicated; protease domain (red); PDZ1 domain (dark blue); PDZ2 domain (yellow); PDB code: 1KY9[35]. C: DegPEc 24-mer (colors as in B); PDB code: 3CS0[36].
Shortly after the publication of our work on DegQLf[34], the biochemical and structural characterization of DegQ fromE.coli(DegQEc) has been reported by another group[42]. DegQ proteins fromLegionellafalloniiandE.colishare an amino acid sequence identity of 41%. Like DegQLf, theE.colihomologue is able to form protease-active higher-order oligomers which are regulated by the conserved molecular switch described above. However, other structural and functional aspects are not shared. In stark contrast to the corresponding variant of DegQLf, DegQEcΔPDZ2 is still able to form proteolytically active 12-mers. Moreover, 6-meric and 24-meric forms, which were absent in all our DegQLppreparations[34], have been observed for DegQEc[42]. Thus, in many ways DegQEcresembles DegPEc, the second chaperone-protease of theE.coliperiplasm. Because expression of thedegQEcgene is not inducible by high temperatures[33], it has been proposed that DegQEchas house-keeping functions, counteracting pH-mediated accumulation of unfolded proteins[42]. In the presence of an excess of unfolded periplasmatic proteins, DegPEcis upregulated to provide the required additional refolding/degradation capacity. LikeLegionellafallonii, many prokaryotes lack a DegP homologue, with DegQ being the only periplasmatic chaperone-protease responsible for protein homeostasis in these species. It is therefore not surprising that DegQLfand DegQEcdiffer in important aspects of oligomerization and function. Future research will reveal if unique features of DegQLfare prototypical for solitary prokaryotic HtrA chaperone-proteases.
3 Conclusion and future perspectives
About 35 years have passed sinceL.pneumophilawas identified as a new human pathogen. However, medical and public interest have increased over the years, and according to the European Work Group forLegionellaInfections (EWGLI), more than 32 000 cases of Legionnaires’ disease resulting in 2 600 deaths have been reported in Europe in the 11 years between 1995 and 2005. Still, it is very likely that the number of cases of Legionnaires’ disease is vastly underreported. In 2009, a study to rigorously analyze the incidence of severe pneumonia caused byLegionellassp. was conducted[43]. Using diagnosis tools specifically aimed to identifyLegionellaspecies, 15 000-30 000 cases of Legionnaires’ disease per year were estimated for Germany alone[43]. Therefore, the associated health-care problem seems to be much more severe than anticipated.
If correctly diagnosed, most cases of Legionnaires’ disease can be treated successfully with fluoroquinolones and macrolides, such as levofloxacin[44]and azithromycin[45]. So far, there are no reports thatLegionellaspp. develop resistance to antibiotic therapy in clinical settings. However, resistance to many clinically relevant drugs including macrolides, quinolones, and rifampicin can be induced in laboratory experiments[46-49]. To counteract emerging resistance, it is therefore vital to develop new drugs, ideally directed against new target proteins ofLegionella. In this respect,LpMip represents an excellent example for structure-guided drug design. The reported Mip structures reveal a detailed picture of the active site that could be targeted by future drugs. Furthermore, complexes with FK506 and rapamycin[15,19]allowed the identification and chemical exploration of a first lead structure[20]. Similar approaches, especially when combined with structure determination of complexes between Mip and inhibitory molecules, hold great promise for the successful development of Mip inhibitors acting as efficient antibiotics. Furthermore, the availability of additional structural data on Mip proteins from different species, in particular inhibitor complexes, is highly desirable to facilitate an in-depth understanding of this protein family.
Over the last years, structural information on new virulence proteins ofLegionellaspp. has slowly been accumulating. Among others, the recently determined structures of DegQ and NFeoB offer unique starting points for the development of innovative antibacterials. Analogous to Mip, in these systems the catalytically active sites represent potential binding sites for inhibitory molecules. However, as both proteins depend on large- and small-scale conformational changes, these could also be targeted. New substances could, for example, interfere with the assembly of active DegQ particles or block the signal transduction from the nucleotide-binding site to the transmembrane region of FeoB. We hope that public availability of structural information on new virulence proteins ofLegionellawill support efforts to shed light on basal mechanisms of pathogenicity and act as a primer for the development of new antibiotics.
Acknowledgements
We thank R. Wrase for help with the preparation of figures for this manuscript.
[1] McDade JE, Shepard CC, Fraser DW, Tsai TR, Redus MA, Dowdle WR. Legionnaires’ disease: isolation of a bacterium and demonstration of its role in other respiratory disease [J]. N Engl J Med, 1977, 297(22): 1197-1203.
[2] Fields BS, Benson RF, Besser RE. Legionella and Legionnaires’ disease: 25 years of investigation [J]. Clin Microbiol Rev, 2002, 15(3): 506-526.
[3] WHO. Legionella and the Prevention of Legionellosis [M]. Geneva: World Health Organization Press, 2007.
[4] Cianciotto NP, Eisenstein BI, Mody CH, Toews GB, Engleberg NC. A Legionella pneumophila gene encoding a species-specific surface protein potentiates initiation of intracellular infection [J]. Infect Immun, 1989, 57(4): 1255-1262.
[5] Leuzzi R, Serino L, Scarselli M, Savino S, Fontana MR, Monaci E, Taddei A, Fischer G, Rappuoli R, Pizza M. Ng-MIP, a surface-exposed lipoprotein of Neisseria gonorrhoeae, has a peptidyl-prolyl cis/trans isomerase (PPIase) activity and is involved in persistence in macrophages [J]. Mol Microbiol, 2005, 58(3): 669-681.
[6] Lundemose AG, Kay JE, Pearce JH. Chlamydia trachomatis Mip-like protein has peptidyl-prolyl cis/trans isomerase activity that is inhibited by FK506 and rapamycin and is implicated in initiation of chlamydial infection [J]. Mol Microbiol, 1993, 7(5): 777-783.
[7] Moro A, Ruiz-Cabello F, Fernndez-Cano A, Stock RP, Gonzlez A. Secretion by Trypanosoma cruzi of a peptidyl-prolyl cis-trans isomerase involved in cell infection [J]. EMBO J, 1995, 14(11): 2483-2490.
[8] Norville IH, O’Shea K, Sarkar-Tyson M, Zheng S, Titball RW, Varani G, Harmer NJ. The structure of a Burkholderia pseudomallei immunophilin-inhibitor complex reveals new approaches to antimicrobial development [J]. Biochem J, 2011, 437(3):413-422.
[9] Fischer G, Bang H, Ludwig B, Mann K, Hacker J. Mip protein of Legionella pneumophila exhibits peptidyl-prolyl-cis/trans isomerase (PPlase) activity [J]. Mol Microbiol, 1992, 6(10): 1375-1383.
[10] Helbig JH, König B, Knospe H, Bubert B, Yu C, Lück CP, Riboldi-Tunnicliffe A, Hilgenfeld R, Jacobs E, Hacker J, Fischer G. The PPIase active site of Legionella pneumophila Mip protein is involved in the infection of eukaryotic host cells [J]. Biol Chem, 2003, 384(1):125-137.
[11] Wagner C, Khan AS, Kamphausen T, Schmausser B, Unal C, Lorenz U, Fischer G, Hacker J, Steinert M. Collagen binding protein Mip enables Legionella pneumophila to transmigrate through a barrier of NCI-H292 lung epithelial cells and extracellular matrix [J]. Cell Microbiol, 2007, 9(2): 450-462.
[12] Wintermeyer E, Ludwig B, Steinert M, Schmidt B, Fischer G, Hacker J. Influence of site specifically altered Mip proteins on intracellular survival of Legionella pneumophila in eukaryotic cells [J]. Infect Immun, 1995, 63(12): 4576-4583.
[13] Cianciotto NP, Fields BS. Legionella pneumophila mip gene potentiates intracellular infection of protozoa and human macrophages [J]. Proc Natl Acad Sci USA, 1992, 89(11): 5188-5191.
[14] Horne SM, Kottom TJ, Nolan LK, Young KD. Decreased intracellular survival of an fkpA mutant of Salmonella typhimurium Copenhagen [J]. Infect Immun, 1997, 65(2): 806-810.
[15] Riboldi-Tunnicliffe A, König B, Jessen S, Weiss MS, Rahfeld J, Hacker J, Fischer G, Hilgenfeld R. Crystal structure of Mip, a prolylisomerase from Legionella pneumophila [J]. Nat Struct Biol, 2001, 8(9): 779-783.
[16] Köhler R, Fanghänel J, König B, Lüneberg E, Frosch M, Rahfeld JU, Hilgenfeld R, Fischer G, Hacker J, Steinert M. Biochemical and functional analyses of the Mip protein: influence of the N-terminal half and of peptidylprolyl isomerase activity on the virulence of Legionella pneumophila [J]. Infect Immun, 2003, 71(8): 4389-4397.
[17] Budiman C, Bando K, Angkawidjaja C, Koga Y, Takano K, Kanaya S. Engineering of monomeric FK506-binding protein 22 with peptidyl prolyl cis-trans isomerase. Importance of a V-shaped dimeric structure for binding to protein substrate [J]. FEBS J, 2009, 276(15): 4091-4101.
[18] Pereira PJ, Vega MC, González-Rey E, Fernández-Carazo R, Macedo-Ribeiro S, Gomis-Rüth FX, González A, Coll M. Trypanosoma cruzi macrophage infectivity potentiator has a rotamase core and a highly exposed alpha-helix [J]. EMBO Rep,2002, 3(1): 88-94.
[19] Ceymann A, Horstmann M, Ehses P, Schweimer K, Paschke AK, Steinert M, Faber C. Solution structure of the Legionella pneumophila Mip-rapamycin complex [J]. BMC Struct Biol, 2008, 8:17.
[20] Juli C, Sippel M, Jäger J, Thiele A, Weiwad M, Schweimer K, Rösch P, Steinert M, Sotriffer CA, Holzgrabe U. Pipecolic acid derivatives as small-molecule inhibitors of the Legionella MIP protein [J]. J Med Chem, 2011, 54(1): 277-283.
[21] Liles MR, Scheel TA, Cianciotto NP. Discovery of a nonclassical siderophore, legiobactin, produced by strains of Legionella pneumophila. J Bacteriol, 2000, 182(3): 749-757.
[22] Cianciotto NP. Iron acquisition by Legionella pneumophila [J]. Biometals, 2007, 20(3-4): 323-331.
[23] Kammler M, Schön C, Hantke K. Characterization of the ferrous iron uptake system of Escherichia coli [J]. J Bacteriol, 1993, 175(19): 6212-6219.
[24] Eng ET, Jalilian AR, Spasov KA, Unger VM. Characterization of a novel prokaryotic GDP dissociation inhibitor domain from the G protein coupled membrane protein FeoB [J]. J Mol Biol, 2008, 375(4): 1086-1097.
[25] Petermann N, Hansen G, Schmidt CL, Hilgenfeld R. Structure of the GTPase and GDI domains of FeoB, the ferrous iron transporter of Legionella pneumophila [J]. FEBS Lett, 2010, 584(4): 733-738.
[26] Köster S, Wehner M, Herrmann C, Kühlbrandt W, Yildiz O. Structure and function of the FeoB G-domain from Methanococcus jannaschii. J Mol Biol,2009, 392(2): 405-419.
[27] Hattori M, Jin Y, Nishimasu H, Tanaka Y, Mochizuki M, Uchiumi T, Ishitani R, Ito K, Nureki O. Structural basis of novel interactions between the small-GTPase and GDI-like domains in prokaryotic FeoB iron transporter [J]. Structure, 2009, 17(10): 1345-1355.
[28] Ash MR, Guilfoyle A, Clarke RJ, Guss JM, Maher MJ, Jormakka M. Potassium-activated GTPase reaction in the G protein-coupled ferrous iron transporter B [J]. J Biol Chem, 2010, 285(19): 14594-14602.
[29] Hung KW, Chang YW, Eng ET, Chen JH, Chen YC, Sun YJ, Hsiao CD, Dong G, Spasov KA, Unger VM, Huang TH. Structural fold, conservation and Fe(II) binding of the intracellular domain of prokaryote FeoB [J]. J Struct Biol, 2010, 170(3): 501-512.
[30] Guilfoyle A, Maher MJ, Rapp M, Clarke R, Harrop S, Jormakka M. Structural basis of GDP release and gating in G protein coupled Fe2+transport [J]. EMBO J, 2009, 28(17): 2677-2685.
[31] Clausen T, Southan C, Ehrmann M. The HtrA family of proteases: implications for protein composition and cell fate [J]. Mol Cell, 2002, 10(3): 443-455.
[32] Ingmer H, Brondsted L. Proteases in bacterial pathogenesis [J]. Res Microbiol, 2009, 160(9): 704-710.
[33] Waller PR, Sauer RT. Characterization of degQ and degS, Escherichia coli genes encoding homologs of the DegP protease [J]. J Bacteriol, 1996, 178(4): 1146-1153.
[34] Wrase R, Scott H, Hilgenfeld R, Hansen G. The Legionella HtrA homologue DegQ is a self-compartmentizing protease that forms large 12-meric assemblies [J]. Proc Natl Acad Sci USA, 2011, 108(26): 10490-10495.
[35] Krojer T, Garrido-Franco M, Huber R, Ehrmann M, Clausen T. Crystal structure of DegP (HtrA) reveals a new protease-chaperone machine [J]. Nature, 2002, 416(6879): 455-459.
[36] Krojer T, Sawa J, Schäfer E, Saibil HR, Ehrmann M, Clausen T. Structural basis for the regulated protease and chaperone function of DegP [J]. Nature, 2008, 453(7197): 885-890.
[37] Jiang J, Zhang X, Chen Y, Wu Y, Zhou ZH, Chang Z, Sui SF. Activation of DegP chaperone-protease via formation of large cage-like oligomers upon binding to substrate proteins [J]. Proc Natl Acad Sci USA,2008, 105(33): 11939-11944.
[38] Kim S, Grant RA, Sauer RT. Covalent linkage of distinct substrate degrons controls assembly and disassembly of DegP proteolytic cages [J]. Cell, 2011, 145(1): 67-78.
[39] Sohn J, Grant RA, Sauer RT. OMP peptides activate the DegS stress-sensor protease by a relief of inhibition mechanism [J]. Structure, 2009, 17(10): 1411-1421.
[40] Krojer T, Sawa J, Huber R, Clausen T. HtrA proteases have a conserved activation mechanism that can be triggered by distinct molecular cues [J]. Nat Struct Mol Biol, 2010, 17(7): 844-852.
[41] Kley J, Schmidt B, Boyanov B, Stolt-Bergner PC, Kirk R, Ehrmann M, Knopf RR, Naveh L, Adam Z, Clausen T. Structural adaptation of the plant protease Deg1 to repair photosystem II during light exposure [J]. Nat Struct Mol Biol, 2011, 18(6): 728-731.
[42] Sawa J, Malet H, Krojer T, Canellas F, Ehrmann M, Clausen T. Molecular adaptation of the DegQ protease to exert protein quality control in the bacterial cell envelope [J]. J Biol Chem, 2011, 286(35): 30680-30690.
[43] von Baum H, Ewig S, Marre R, Suttorp N, Gonschior S, Welte T, Lück C, Competence Network for Community Acquired Pneumonia Study Group. Community-acquired Legionella pneumonia: new insights from the German competence network for community acquired pneumonia [J]. Clin Infect Dis, 2008, 46(9): 1356-1364.
[44] Yu VL, Plouffe JF, Pastoris MC, Stout JE, Schousboe M, Widmer A, Summersgill J, File T, Heath CM, Paterson DL, Chereshsky A. Distribution of Legionella species and serogroups isolated by culture in patients with sporadic community-acquired legionellosis: an international collaborative survey [J]. J Infect Dis, 2002, 186(1): 127-128.
[45] Plouffe JF, Breiman RF, Fields BS, Herbert M, Inverso J, Knirsch C, Kolokathis A, Marrie TJ, Nicolle L, Schwartz DB. Azithromycin in the treatment of Legionella pneumonia requiring hospitalization [J]. Clin Infect Dis, 2003, 37(11): 1475-1480.
[46] Dowling JN, McDevitt DA, Pasculle AW. Isolation and preliminary characterization of erythromycin-resistant variants of Legionella micdadei and Legionella pneumophila [J]. Antimicrob Agents Chemother, 1985, 27(2): 272-274.
[47] Jonas D, Engels I, Hartung D, Beyersmann J, Frank U, Daschner FD. Development and mechanism of fluoroquinolone resistance in Legionella pneumophila [J]. J Antimicrob Chemother, 2003, 51(2): 275-280.
[48] Moffie BG, Mouton RP. Sensitivity and resistance of Legionella pneumophila to some antibiotics and combinations of antibiotics [J]. J Antimicrob Chemother, 1988, 22(4): 457-462.
[49] Nielsen K, Hindersson P, Hoiby N, Bangsborg JM. Sequencing of the rpoB gene in Legionella pneumophila and characterization of mutations associated with rifampin resistance in the Legionellaceae [J]. Antimicrob Agents Chemother, 2000, 44(10): 2679-2683.
