
3,5-O-二咖啡酰奎宁酸的合成*
2012-01-11 11:57张亚梅赵法兴
山东第一医科大学(山东省医学科学院)学报 2012年5期
张亚梅 刘 梅 赵法兴
(江西中医药高等专科学校药学系,江西 抚州 344000)
3,5-O-二咖啡酰奎宁酸及其类似物(DCQAs)是由2分子咖啡酸与1分子奎宁酸通过3,5-O-二酯键连接构成的天然多酚类化合物,在我国传统中药苍耳、金银花、白花刺参、灯盏细辛、款冬花、朝鲜蓟中均有分布,具有抗氧化、抗菌、抗组胺、抗肝纤维化等多种生物活性[1]。最新研究证明其能够抑制癌蛋白E6和E6AP,同时抑制宫颈癌细胞的增殖,具有治疗HPV感染的宫颈癌作用[2]。尽管DCQAs在多种中草药中有分布,但均因含量低,分离困难,制约了其进一步的药效和临床研究,因此,探索人工合成该类化合物的工作迫在眉睫。有研究人员对其进行了全合成的研究,但产率较低[3],为提高产率,降低合成成本,本人在前人工作的基础上对3, 5-O-二咖啡酰奎宁酸及其类似物的合成进行研究。现将合成路线总结如下。
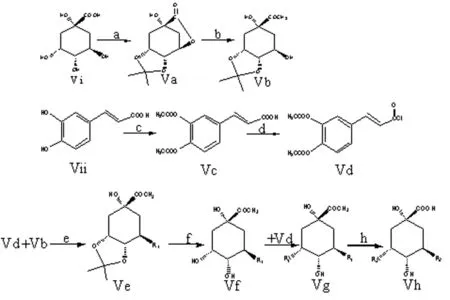
上述结构中:

1 材料与方法
1.1 材料
1.1.1仪器 旋转薄膜蒸发仪(R502型),万分之一电子天平(Satorius型),超声波清洗器(KQ-500E型),全自动型鼓风干燥箱(ZRD-5030型),循环水式多用真空泵(SHB-B95型),三用紫外仪(ZF-2型),显微熔点测定仪(X-6型),四极杆质谱仪(HP 5988A型), ESI-MS(Esquire 3000plus型),红外光谱仪(Nicolet AVATAR 360 FT-IR型), 超导核磁共振仪(Brucker AM-400型)等。
1.1.2试药 奎宁酸、咖啡酸(ACROS ORGANICS公司提供)。其他试剂均为分析纯。
1.2 方法
1.2.13,4-丙酮叉奎宁酸-5-内酯(Va)的制备[4]
将15.0 g奎宁酸加到400 ml通有氯化氢气体的丙酮溶液(含有氯化氢约4.9%)中,室温持续搅拌30h,TLC显示反应完全,停止反应。反应瓶中通入干燥氨气,至pH=9.0,过滤,滤液浓缩,干燥后进一步用硅胶柱层析洗脱纯化,洗脱剂为石油醚:氯仿=1∶1,收集洗脱液浓缩后析出白色针状结晶,重结晶后得化合物Va。
1.2.23,4-丙酮叉奎宁酸甲酯(Vb)的制备[5]
将0.92g(40.0 mmol)的金属钠分批加入到200 ml的无水甲醇中,待钠反应完全,且体系温度降至室温后,加入6.78 g(30 mmol)的中间体Va,室温(27 ℃)持续搅拌6.5 h,TLC显示反应完全,停止反应。冰浴冷却30 min后,边搅拌边滴加冰醋酸,将pH值调至中性,减压蒸除甲醇,瓶内残余物为白色固体,加入150 ml的二氯甲烷,将固体滤去,并用二氯甲烷洗涤数次,滤液用饱和氯化钠溶液30 ml洗涤,有机层用无水硫酸镁干燥过夜。过滤,浓缩滤液,进一步用硅胶柱层析分离纯化,洗脱剂为石油醚:丙酮=1∶3、1∶1.5、1∶1,得到无色油状物。
1.2.33,4-O-二乙酰咖啡酸的制备Vc[6]
将2.69g(14.96 mmol)的咖啡酸溶解于40 ml的无水吡啶中,冰水浴冷却10 min后,滴加10滴甲酸,冰水浴下搅拌8小时,反应完全。低温减压蒸除过量的吡啶,残余物用稀盐酸酸化至pH=3,加入二氯甲烷提取,有机层用饱和氯化钠溶液洗涤,滤液浓缩,粗品用乙酸乙酯重结晶,得白色固体。
1.2.43,4-O-二乙酰咖啡酰氯的制备Vd[7]
将0.78g(6.15 mmol)的草酰氯、10 ml无水乙醚和5滴无水DMF加入到干燥后的中间体Vc1.474g(2.35 mmol)中,室温(20 ℃)搅拌,溶液逐渐澄清,约4 h后,反应基本完全,进一步分离纯化后得白色固体Vd。
1.2.55-0-(3,4-O-二乙酰咖啡酰基)-3,4-丙酮叉奎宁酸甲酯的制备[7]
将中间体3,4-丙酮叉奎宁酸甲酯(0.344g,1.42 mmol)与碘化钾(22.6 mg,0.136 mmol)溶解于DMF中,50 ℃搅拌15分钟,加入中间体化合物Vd(0.519 g,1.85 mmol),13 ℃下搅拌反应1.5 h。TLC跟踪检测反应完全,减压蒸除溶剂,所得粗产物经柱层析纯化,得到白色固体Ve。
1.2.65-0-(3,4-O-二乙酰咖啡酰基)-3,4-丙酮叉奎宁酸甲酯脱丙酮叉保护基团Vf
将0.273 g(0.56 mmol)的中间体化合物Ve避光,密闭保存放置,用TLC跟踪检测,15天后原料点消失,得到目标产物Vf。
1.2.73,5-0-(3,4-O-二乙酰咖啡酰基)-奎宁酸甲酯的制备Vg[7]
向反应瓶中加入中间体Vd(0.267 g,0.95 mmol),二环己基碳二亚胺(0.29 g,1.4 mmol),无水二氯甲烷15 ml,在室温下搅拌30 min后,加入反应中间体Vf(0.222 mg,0.50 mmol),4-二甲氨基吡啶(0.18 mg,0.15 mmol),整个溶液在室温下反应过夜,抽滤除去不溶物,蒸去溶剂,脱溶得到白色固体,柱层析纯化,得到白色无定型粉末。
1.2.83,5-0-二咖啡酰基-奎尼酸制备Vh[7]
在二颈瓶中加入化合物Vg(0.142 g,0.21 mmol),加入5mL甲醇-四氢呋喃-水体系(3∶2∶2),再滴加5滴1.5 mol/LHCI,加热回流3 h,由TLC检测得到产物和原料不再变化为止,冷却至室温,用二氯甲烷萃取,合并有机层用大量水洗,饱和食盐水洗涤,无水硫酸钠干燥。进一步柱层析得到目标化合物Vh。
2 结 果
2.1 化合物表征
利用1HNMR、13CNMR、DEPT、HRMS等波谱技术并对照原料咖啡酸和奎尼酸的谱图,结合文献[4-7]对合成的中间体及其目标化合物进行了结构鉴定。目标化合物Vh,ESI/TOF/MS,m/z:515[M-H]-,353[M-162-H]-,191[M-162-162-H]-,135,推测其分子式为 C25H24O12。IR(KBr,cm-1)中:3423,1693,1606,1522,l447,1369,1281,1182,1118,980,855,813,600等数据显示有羧基、酚羟基、不饱和双键及酯基结构的吸收。在1HNMR(D2O,300MHz)中,δ(ppm):δH7.44(t,2H,H-7′,H-7′′),6.72(d,2H,H-5′,H-5′′),6.23(d,1H,H-8′),6.14(d,1H,H-8′′)显示有两个咖啡酰基取代基的存在;7.00( 2H, 2dd, J=2.2, 8.4Hz, 6′-H×2),6.78, 6.77(2H, 2d, J= 8.4 Hz, 5′-H×2),示2个苯环均为1′,3′,4′取代;5.28(m,1H,H-5′′),5.16(d,1H,H-3),3.902(m,IH,H-4),1.94-2.20(m,4H,H-2,H-6)显示有奎尼酸的存在。
13CNMR(D2O,300MHz)δ(ppm):171.47,171.21(C-9′,C-9′′),166.94.(C-5′),150.57(C-5′′),149.65(C-4′),148.84(C-4′′),146.82(C-7′),139.19(C-7′′),129.58(C-1′),129.50(C-1′′),125.32(C-2′,C-2′′),118.76(C-3′,C-3′′),117.63(C-8′),117.20(C-8′′),116.97(C-6′),116.02(C-6′′)印证有两个咖啡酰基取代基的存在;177.04(C-7,COOH),78.44(C-l),77.12(C-4),74.61(C-5).73.17(C-3),48.58[45.73](C-2),38.76(C-6)印证了奎尼酸的存在。其它各中间体的结构鉴定数据见表1至3。
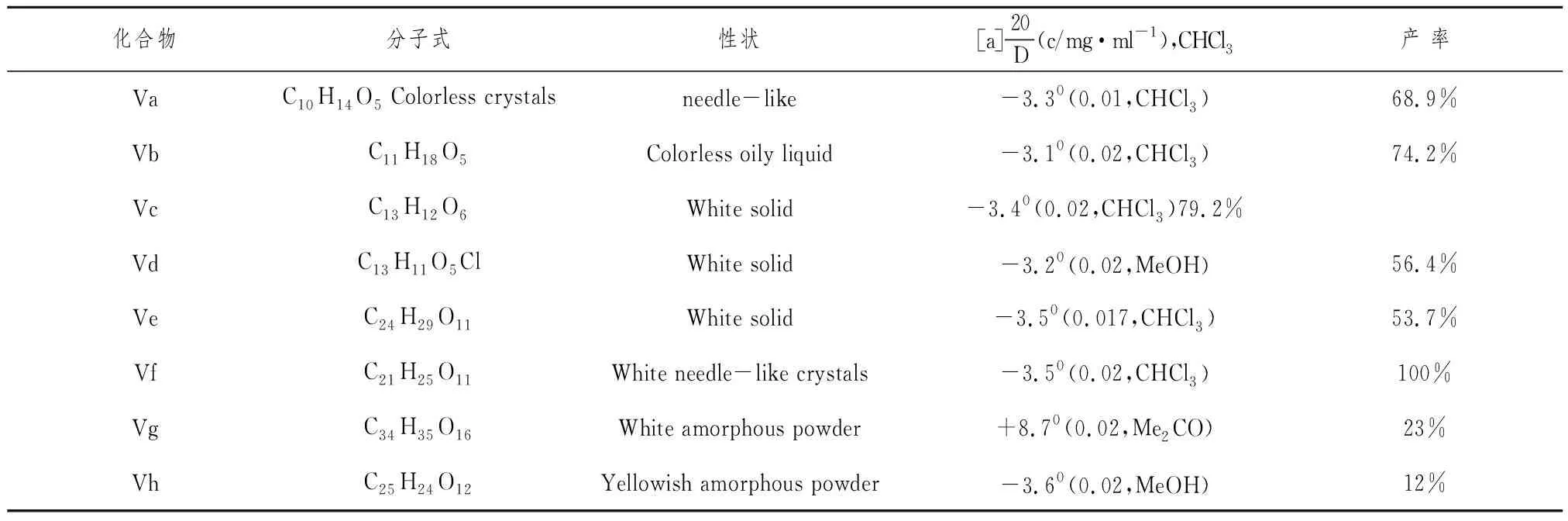
表1 化合物Va~Vg的物理数据

表2 合成化合物的质谱数据

表3 合成化合物的1H NMR/Mp数据
3 讨 论
根据目的化合物3,5-O-二咖啡酰奎尼酸的结构特点,本文选择咖啡酸和奎尼酸作为合成原料。由于奎宁酸含有四个羟基和一个羧基,咖啡酸也含有两个活泼的酚羟基,因此,设计路线时,考虑与选择这些活泼官能团的保护方法及保护与脱保护的反应条件,是成功实施全合成的关键所在。在奎尼酸的选择性保护过程中,在文献[4]报道的基础上[4],提高氯化氢浓度,并降低温度能够使反应率提高到68.9%。而在成酯反应过程中,以吡啶作为有机碱进行反应,不仅可中和反应形成的氯化氢,还可活化酰氯,提高反应率。在脱丙酮叉保护基团的过程中,采用文献[5]中的方法,将样品避光,密闭保存,放置,2周后,丙酮叉自动脱去,2个酯键未受影响。在进行咖啡酸的酰氯反应中,室温条件下改用乙醚作溶剂反应完全,副产物较少。
[1] 刘波.杏香兔耳风活性成分研究[D〗.北京:首都师范大学化学系,2005.
[2] 李成晟,祝志安.二咖啡酰奎宁酸药理实验研究进展[J〗.医学综述,2004, 10:249-251.
[3] 程伟贤,陈鸿雁,张义平,等.山牡荆的化学成分研究[J].天然产物研究与开发,2007, 19: 244-246
[4] 田景奎.珍珠菜属两种药用植物化学成分的研究[D].北京:中国医学科学院,1997.
[5] 吴小瑛.1,5-二-O-咖啡酰奎宁酸及其类似物的全合成[D].北京:中国协和医科大学,2000.
[6] 刑其毅,基础有机化学[M].北京:高等教育出版社,1980,466.
[7] 王葆仁,有机合成反应[M].北京:科学出版社,1981,319-347
猜你喜欢
分析测试学报(2020年4期)2020-05-09
吉林农业(2019年6期)2019-06-11
发酵科技通讯(2018年2期)2018-07-06
中成药(2017年10期)2017-11-16
中成药(2017年4期)2017-05-17
中国洗涤用品工业(2017年2期)2017-04-16
当代化工研究(2016年2期)2016-03-20
中国洗涤用品工业(2016年2期)2016-02-28
应用化工(2014年5期)2014-08-08
中成药(2014年9期)2014-02-28
