
磷钨酸光催化降解直接大红4BE溶液的研究
2011-12-20 09:11李克斌西安理工大学西北水资源与环境生态教育部重点实验室陕西西安70048西北大学化学与材料科学学院合成与天然功能分子化学教育部重点实验室陕西西安70069
中国环境科学 2011年6期
魏 红,李克斌,赵 锋,张 涛,李 娟 (.西安理工大学西北水资源与环境生态教育部重点实验室,陕西 西安 70048;.西北大学化学与材料科学学院,合成与天然功能分子化学教育部重点实验室,陕西 西安 70069)
磷钨酸光催化降解直接大红4BE溶液的研究
魏 红1,李克斌2*,赵 锋2,张 涛2,李 娟1(1.西安理工大学西北水资源与环境生态教育部重点实验室,陕西 西安 710048;2.西北大学化学与材料科学学院,合成与天然功能分子化学教育部重点实验室,陕西 西安 710069)
以磷钨酸(PW12)为催化剂,对偶氮染料直接大红4BE进行均相光催化降解,考察了PW12用量、染料初始浓度对反应的影响,并对反应机理进行了探讨.结果表明,PW12能够有效光催化降解直接大红4BE.PW12用量≤600mg/L时,直接大红4BE的降解随PW12用量的增加明显加快, 光解过程符合表观一级反应动力学.当pH2.0、直接大红4BE初始浓度为50mg/L、PW12用量为600mg/L时,其光催化降解效果最佳,对应的一级表观反应速率常数k为0.1164min-1.直接大红4BE初始浓度在50~150mg/L范围内,PW12对其光催化降解速率随染料浓度的增加而减小.结合直接大红4BE的循环伏安图,以及不同实验条件下PW12光催化直接大红4BE的UV-vis光谱图,进行的机理探讨结果表明,实验条件下,直接大红4BE的PW12光催化脱色过程是•OH氧化, PW12*–直接大红4BE复合物内的电子转移,以及杂多蓝PW12(e-)还原共同作用的结果,其中⋅OH氧化脱色起主导地位.
磷钨酸;均相光催化;直接大红4BE;反应机理
偶氮染料是一类广泛使用的人工合成染料,由于其本身的毒性、降解产物的潜在致癌作用,对环境存在极大的危害[1].目前,偶氮染料废水的处理方法主要包括吸附、生物、氧化和还原降解等[2].高级氧化技术如 TiO2和多金属氧酸盐(POM)光催化体系,能够有效将氧化和还原作用结合起来,其光催化降解有机污染物一直是环境污染控制领域中的研究热点之一[3-5].关于 POM尤其是 H3PW12O40(PW12)的应用,近些年备受关注,其光催化降解的污染物包括有机氟[6-7]、氯酚类[8]、丙酮[4]等化合物.PW12催化降解水中有机污染通常采用均相和非均相2种形式.鉴于PW12的水溶性,均相催化有利于催化剂与污染物充分接触,有效避免非均相催化中催化剂对光的散射和透光率低的弱点.但均相催化存在催化剂难回收、多酸比表面较低等问题,这些可以通过负载PW12解决.相对TiO2光催化,目前PW12光催化降解偶氮染料还缺乏系统的研究[9].本试验以PW12为催化剂,研究其对偶氮染料直接大红4BE的均相光催化降解性能,考察反应的影响因素,并探讨光催化氧化和还原机理.
1 材料与方法
1.1 实验材料
NaOH、NaClO4、H3PW12O40、异丙醇、KCl、磷酸、硼酸、冰醋酸均为分析纯;直接大红4BE,分子式:C34H26N6(SO3Na)2,工业纯;蒸馏水.
1.2 实验仪器
754PC紫外–可见分光光度计(上海光谱仪器有限公司);pHs-25数显酸度计(上海虹益仪器仪表有限公司);中压汞灯(500W,开封宏兴科教仪器厂),光强度测定按照文献[10],采用三草酸合铁酸钾化学露光计,以波长256nm下的光辐射表示,其值为 4.96×10-7Ein/s;电化学工作站(上海辰华仪器公司);CL-200型磁力搅拌器;光反应器(开封宏兴科教仪器厂).
1.3 实验方法
1.3.1 直接大红4BE氧化还原峰电位测定 参照文献[11-12],通过循环伏安法在电化学工作站上进行.测定时采取三电极体系,玻碳电极为工作电极,参比电极为饱和甘汞电极,铂丝电极作为辅助电极.待测液包括1.0×10-2mol/L的染料储备液5.0mL, 1.0mol/L KCl溶液5.0mL作为支持电解质,由磷酸、冰醋酸和硼酸组成的缓冲溶液5.0mL (pH2.0).在进行循环伏安扫描前,向反应液中通入N2排除溶解氧.扫描速率20~200mV/s,扫描范围-1500~900mV.
1.3.2 PW12光催化降解直接大红4BE 移取一定体积5.0g/L的直接大红4BE储备液至500mL容量瓶中,加入 PW12水溶液, 通过 1.0mol/L的 NaOH和HClO4水溶液调节体系pH值,定容.打开光源,光辐射稳定后(10min),将反应液转入反应器,每隔一段时间取样,在波长572nm处测定样品浓度C,以C/C0对t作图分析.其中, C0和C分别为0和t时刻直接大红4BE的浓度,mg/L.反应过程中鼓入空气时,通过一流量为2.0L/min的空气泵连接一导管深入反应器底部.
2 结果与讨论
2.1 PW12用量的影响
实验考察了 pH2.0、直接大红 4BE浓度为50mg/L、PW12用量为0~1000mg/L条件下,直接大红4BE的光降解过程(图1).
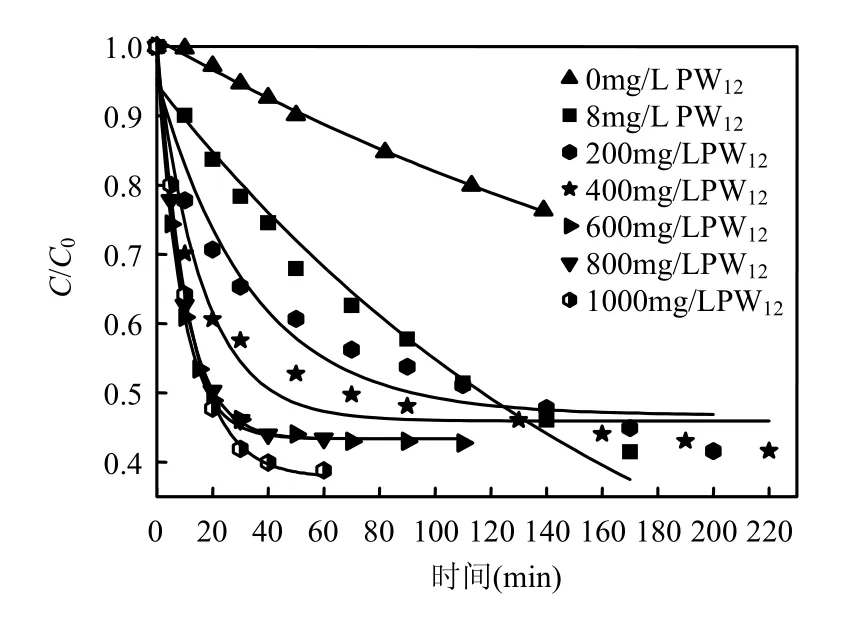
图1 PW12 用量对直接大红4BE光解过程的影响Fig.1 Effect of PW12 concentration on 4BE photo-degradation
由图1可知, PW12用量≤600mg/L内,直接大红4BE的光解随PW12用量的增加明显加快;当PW12用量依次为 8,200,400,600mg/L时,反应70min, C/C0从 0.873依次降低为 0.578, 0.538, 0.481和 0.434;当 PW12用量>600mg/L时,增加PW12用量,反应速率变化不明显,但直接大红4BE的最终脱色程度增大,反应 60min,C/C0从600mg/L PW12对应的 0.434降低为 1000mg/L PW12对应的0.388.
采用一级反应动力学方程:C/C0=exp(-kt)对图1进行非线性拟合.结果表明,PW12光催化降解直接大红4BE符合一级反应动力学(R2>0.97).由不同PW12用量与相对应的表观一级反应速率常数(k)的关系可见(图2),PW12用量为600mg/L时,k值达到最大,为0.1164min-1.
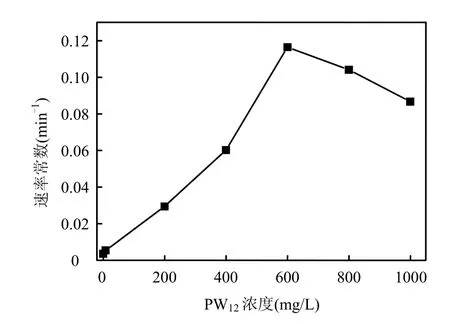
图2 一级反应速率常数与PW12 浓度的关系Fig.2 Relationship of the first-order reaction rate constant and PW12concentration
2.2 直接大红4BE初始浓度的影响
图3为当pH 2.0、PW12用量600mg/L、直接大红4BE的初始浓度分别为50,100,150mg/L时的光催化降解情况.
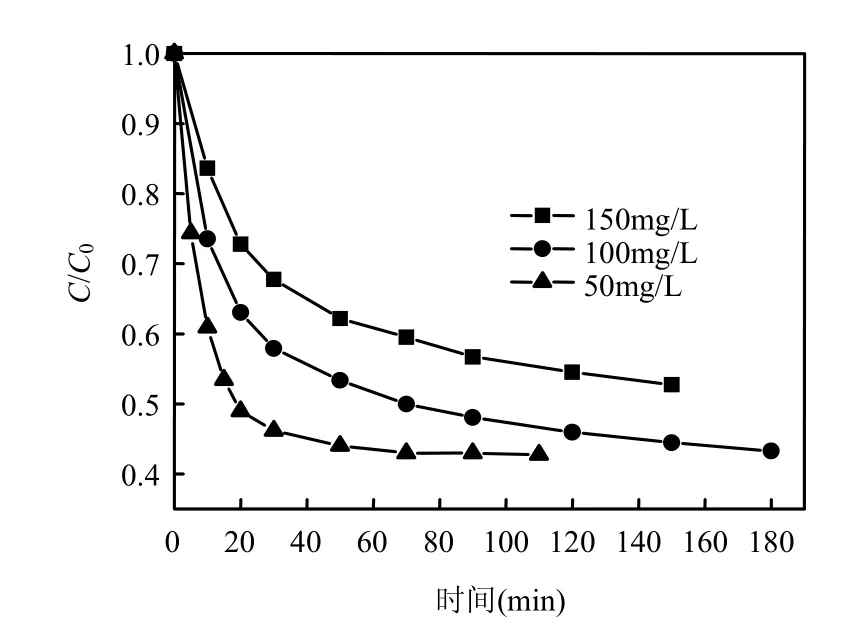
图3 直接大红4BE初始浓度对其光解效果的影响Fig.3 Effect of 4BE initial concentration on itsphotocatalytic degradation
由图3可见,直接大红4BE的光解速度随其初始浓度的增加逐渐减小.按照一级反应动力学方程对图 3进行非线性拟合(R2>0.98),直接大红4BE初始浓度分别为50,100,150mg/L时, k分别为0.1164, 0.1096, 0.0856min-1.
直接大红4BE初始浓度对其催化光解速率
的影响一方面可能是由于滤光效应所致,另一方可能是由于染料在光解过程中会产生许多中间产物,中间产物与染料分子存在竞争氧化[13].实验测得pH2.0时,PW12水溶液的特征吸收峰在波长259nm,直接大红4BE初始浓度从50mg/L增加到150mg/L, A259nm由0.284增加到0.851,在光强度一定时,PW12吸收的紫外光随直接大红4BE初始浓度增加而减少,产生激发态杂多酸 PW12*的量也相应减少,因此直接大红4BE的光催化性能随初始浓度的升高有所降低.
2.3 PW12光催化降解直接大红4BE的机理
2.3.1 PW12光催化降解反应过程 首先 PW12在UV辐照下得到激发态杂多酸PW12*[14][式(1)], PW12*的氧化还原电位为2.63V[15],能够氧化直接大红4BE、溶剂水[16-17][式(2)~式(3)].前者作用的结果产生了染料阳离子自由基,并进一步发生降解;后者作用的结果是产生了非选择性强氧化剂⋅OH, ⋅OH氧化染料使其降解[式(4)].

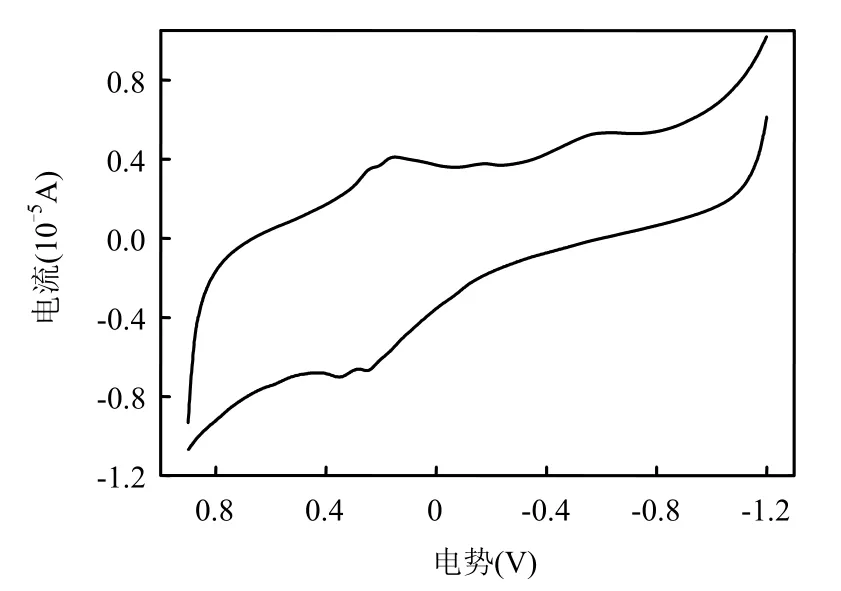
图4 直接大红4BE的循环伏安图Fig.4 Cyclic voltammogram graph of 4BE
在 PW12光催化氧化降解有机污染物的同时,PW12形成单电子还原态-杂多蓝 PW12(e-)[式(2)~式(3)],杂多蓝 PW12(e-)的特征吸收在波长752nm[18],其相对标准氢电极的氧化还原电位为0.221V[19].直接大红4BE的还原电位通过循环伏安法测得为 0.152V(图 4),相对标准氢电极的氧化还原电位为 0.393V[20],0.221V<0.393V,可见PW12(e-)能够还原降解直接大红4BE.
当pH 2.0时, UV辐射600mg/L的PW12与0.13mol/L的异丙醇溶液,生成杂多蓝 PW12(e-).然后,将杂多蓝与酸性大红4BE在避光下混合、密封,混合液的UV-vis光谱的变化情况见图5.
图5 杂多蓝与直接大红4BE暗反应UV-vis光谱
Fig.5 UV-vis spectra of the mixture 4BE and reduced PW12
由图 5可见,杂多蓝 PW12(e-)使酸性大红4BE在波长572nm附近的吸收减弱,偶氮键断裂.同时波长752nm的吸收也不断降低,表明杂多蓝PW12(e-)被氧化复原.
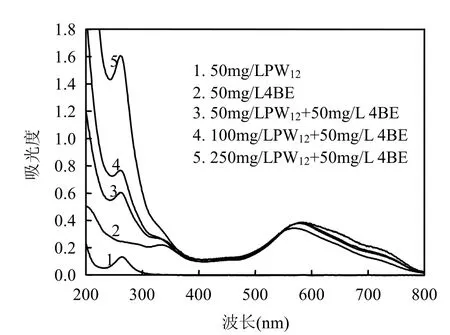
图6 无UV辐照时,PW12用量对直接大红4BE UV-vis吸收光谱的影响Fig.6 Effect of PW12 concentration on the UV-vis spectra of 4BE without UV irradiation
2.3.2 直接大红4BE与PW12的相互作用 PW12水溶液在波长>350nm范围内无吸收,而图6显示,随着PW12用量的增加,直接大红4BE在可见光区的最大吸收从波长569nm红移到波长580nm,直接大红4BE与PW12混合溶液存在等吸收点.这表明pH2.0时,直接大红4BE和PW12相互作用形成复合物[式(5)].

2.3.3 异丙醇和溶解氧对 PW12光催化直接大红 4BE的影响 实验考察了 pH2.0、直接大红4BE初始浓度为50mg/L、PW12用量600mg/L、异丙醇浓度为0.13mol/L时,异丙醇对PW12光催化降解直接大红4BE的影响(图7).

图7 几种实验条件下直接大红4BE的光解情况Fig.7 Photodegradation of 4BE under several experimental conditions
由图7可见,向PW12光催化直接大红4BE体系中加入⋅OH捕获剂异丙醇(IS)[21]后,反应前20min,直接大红4BE脱色速率受到抑制(C/C0为0.93,高于无异丙醇时的 0.49),随后脱色过程加快.反应前20min内,异丙醇对脱色速率的抑制,表明⋅OH是PW12催化降解直接大红 4BE的重要途径之一.20min后反应加速应该是由于杂多蓝 PW12(e-)还原直接大红4BE使其快速脱色[式(6)] .
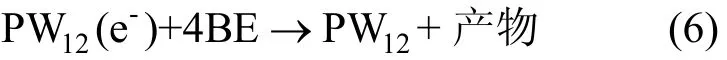
由图8可见,反应10min时,体系在波长752nm处的吸收迅速增加;10~20min范围,直接大红4BE在波长580nm处的吸收变化不大;继续反应,直接大红4BE在波长580nm处的吸收不断减小,同时杂多蓝PW12(e-)的特征吸收也在减小.这可能是由于反应体系中加入异丙醇后促使杂多蓝PW12(e-)生成,随后杂多蓝PW12(e-)又与直接大红4BE发生[式(6)]反应,同时使染料还原脱色.

图8 异丙醇对PW12光催化4BE时UV-vis图谱的影响(不鼓入空气)Fig.8 Effect if isopropanol on UV-vis spectra of the 4BE photocatalytic degradation without bubbling air
为进一步了解杂多蓝 PW12(e-)对直接大红4BE的还原脱色作用,向PW12催化降解直接大红4BE反应体系中鼓入空气.文献报道氧气与PW12(e-)发生式(7)~式(10)的反应[22].
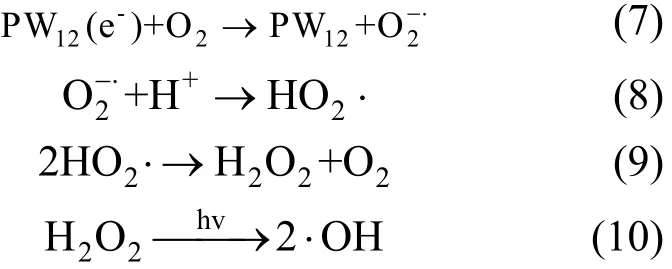
如果存在杂多蓝 PW12(e-)对直接大红 4BE的还原脱色作用,那么反应体系中鼓入空气将会降低直接大红4BE的光解速率.由图7可见,通入空气后PW12光催化降解直接大红4BE的初始速度稍有减缓,但最终脱色率反而提高.可见,PW12光催化降解直接大红4BE过程中存在还原作用.从暗反应(图5)估计,杂多蓝PW12(e-)对直接大红4BE的初始还原脱色速率约为 1.01mg/(L⋅min),而相同PW12和染料浓度下的催化光解初始速率约为5.82mg/(L⋅min)(图2),因此杂多蓝的还原脱色不是本实验条件下的主要脱色过程.
同时,图7表明式(7)和式(8)产生的氧化物种对染料的初始脱色贡献不大.这也可从它们的氧化还原电位的相对大小看出.HO2•/O2,O2•-/O2的氧化还原电位分别为-0.05V和-0.33V[23],而直接大红的氧化电位 0.492V.不过,HO2•、O2•-活性氧自由基可能会降解染料降解过程产生的某些中间体,因此会提高染料最终脱色率.

图9 鼓入空气对4BE催化光解UV-vis光谱的影响Fig.9 UV-vis absorption spectra photocatalytic degradation in the presence of oxygen
比较图9a与图8,发现反应过程中无杂多蓝PW12(e-)的特征吸收峰,溶解 O2加速了还原态杂多酸的复原[式(7)].另外,图9b显示UV-vis光谱在波长260nm附近的吸收随反应进行先增加,后又减小.这可能与反应过程中芳香胺的生成和消失有关.一般认为偶氮染料还原降解过程中会在240~350nm出现与芳胺有关的吸收峰,而芳胺极易被氧化[24].
综上分析,PW12光催化降解直接大红4BE时,反应体系中发生了以下反应:⋅OH对直接大红4BE的氧化而引起的降解; PW12与直接大红4BE之间形成复合物,UV辐照下光激发的PW12*与直接大红4BE发生复合物内电子转移氧化;杂多蓝PW12(e-)对直接大红4BE的还原,在这3个反应中,⋅OH的氧化起主导作用.
3 结论
3.1 PW12能够有效催化光解直接大红 4BE,反应符合表观一级反应动力学.PW12用量为600mg/L时,k值达到最大,为0.1164min-1.
3.2 直接大红4BE的初始浓度在50~150mg/L范围,降解速率随着直接大红4BE初始浓度的增加而降低.
3.3 实验条件下,直接大红4BE的PW12光催化脱色过程是⋅OH氧化、激发态PW12*与直接大红4BE复合物的电子转移氧化,以及单电子还原态-杂多蓝PW12(e-)对直接大红4BE还原共同作用的结果.在这3个反应中,⋅OH的氧化起主导作用.
[1] Forgacs E, Cserháti T, Oros G. Removal of synthetic dyes from wastewaters: A review [J]. Environ Intern, 2004,30:953-971.
[2] Martńez-Huitle C A, Brillas E. Decontamination of wastewaters containing synthetic organic dyes by electrochemical methods: A general review [J]. Applied Catalysis B, 2009,87:105-145.
[3] 郑怀礼,彭德军,李 宏,等.光助Fenton催化氧化反应降解孔雀石绿试验研究 [J]. 光谱学与光谱分析, 2007,27(5):l006-1009.
[4] 邓 谦,吕晓萌,蔡铁军,等.磷钨酸表面修饰 TiO2光催化降解空气污染物 [J]. 中国环境科学, 2005,25(3):375-379.
[5] 苏营营,于艳卿,杨沛珊,等.纳米TiO2/硅藻土光催化降解蒽醌染料废水的研究 [J]. 中国环境科学, 2009,29(11):1171-1176.
[6] 黄 丽,陈 悠,董文博,等.磷钨酸光催化六氟苯脱氟的研究[J]. 环境科学, 2006,27(8):1501-1507.
[7] Hisao Hori, Etsuko Hayakawa, Hisahiro Einaga, et al. Decomposition of environmentally persistent perfluoroctanoic acid in water by photochemical approaches [J]. Environ. Sci. Technol., 2004,38:6118-6124.
[8] Evagelia Androulaki,Anastasia Hiskia,Dimitra Dimotikia, et al. Light induced elimination of mono and polychlorinated phenol from aqueous solution by PW12O403-. The case of 2,4,6-trichlorophenol [J]. Environ. Sci. Technol., 2000,34:2024- 2028.
[9] 李松田,吴春笃,闫永胜,等.杂多酸光催化降解有机污染物 [J].化学进展, 2008,20(5):690-697.
[10] 康锡惠,刘梅清.光化学原理与应用 [M]. 天津大学出版社, 1995:148-153.
[11] Vinod Kumar Gupta, Rajeev Jain, Shaily Varshney. Electrochemical removal of the hazardous dye reactofix red 3BFN from industrial effluents [J]. Journal of Colloid and Interface Science, 2007,312:292-296.
[12] Eleonora-Mihaela Ungureanu, Alexandru C Razus, Liviu Birzan, et al. Electrochemical study of azo-azulene compounds [J]. Electrochimica, 2008,53:7089-7099.
[13] Zhong Jun-bo, Ma Di, Zhao Hong, et al. Kinetic study on photocatalytic degradation of reactive orange 5solution with phosphotungstic acid [J]. J Mol Catal A: Chemical, 2008,283: 93-98.
[14] Andrea Maldotti, Alessandra Molinari, Rossano Amadelli. Photocatalysis with organized systems for the oxofunctionalization of hydrocarbons by O2[J]. Chem. Rev., 2002,102:3811-3836.
[15] 杨 曦,余 刚,孔令仁,等.酸性红3B的杂多酸光催化降解动力学 [J]. 环境科学, 2002,23(3):40-43.
[16] Qu Ping, Zhao Jincai, Shen Tao, et al. TiO2assisted photodegradation of dyes: a study of two competitive primary process in the degradation of RB in an aqueous TiO2colloidal solution [J]. Journal of Molecular Catalysisi A: Chemical, 1998,129:257-268.
[17] Anastasia Hiskia, Athanasius Mylonas, Despina Tsipi, et al. Photocatalytic degradation of lindane in aqueous solution [J]. Pestic.Sci.,1997,50:171-174.
[18] Craig L Hill, Donald A Bouchard, Miryam Kadkhodayan, et al. Catalytic photochemical oxidation of organic substrates by polyoxometalates. Picosend spectroscopy, hotochemistry, and structural properties of charge-transfer complexes between heteropolytungstic acids and dipolar organic compounds [J]. J. Am. Chem. Soc., 1988,110(16):5471-5479.
[19] Gkika E, Troupis A, Hiskia A, et al. Photocatalytic reduction and recovery of mercury by polyoxometalates [J]. Environ. Sci. Technol., 2005,39:4242-4248.
[20] 傅献彩,沈文霞,姚天扬.物理化学 [M]. 北京:高等教育出版社, 1996:602.
[21] Ruya R O, John L F. Kinetic probes of the mechanism of polyoxometalate-mediated photocatalytic oxidation of chlorinated organics [J]. J. Phys. Chem. B, 2000,104:9444-9448.
[22] Patrick Mazellier, Michele Bolte. 3-chlorophenol elimination upon excitation of dilute iron (Ⅲ ) solution: evidence for the only involvement of Fe (OH)2+[J]. Chemosphere, 2001,42:361-366.
[23] Hiskia A, Papaconstantinou E. Photocatalytic oxidation of organic compounds by polyoxometalates of molybdenum and tungsten [J]. Catalyst regeneration by dioxygen, 1992,31(2):163- 167.
[24] Pinheiro H M, Touraud E, Thomas O. Aromatic amines from azo dye reduction: status review with emphasis on direct UV spectrophotometric detection in textile industry wastewaters [J]. Dyes and Pigments, 2004,61:121-139.
Photocatalytic degradation of azo-dye direct red 4BE aqueous solution catalyzed by phosphatotungstic acid.
WEI Hong1, LI Ke-bin2*, ZHAO Feng2, ZHANG Tao2, LI Juan1(1. Key Laboratory of Northwest Water Resources, Environment and Ecology, Ministry of Education, Xi’an University of Technology, Xi’an 710048, China;2. Key Laboratory of Synthetic and Natural Functional Molecule Chemistry, Ministry of Education,School of Chemistry and Material Science, Northwest University, Xi'an 710069, China). China Environmental Science, 2011,31(6):921~926
The photodegradation of azo-dye direct red 4BE (4BE) in aqueous solution by phosphatotungstic acid (PW12) as homogeneous catalyst was studied in a batch photoreactor. The parameters such as the concentration of 4BE and PW12were investigated, and the reaction mechanism involved was discussed. 4BE was decolorized effectively in the presence of phosphatotungstic acid under UV irradiation. When PW12concentration was less than 600mg/L, the photocatalytic decolonization rate of 4BE increased along with the increase of PW12concentration. The process of 4BE degradation followed a pseudo-first-order kinetics, and a maximum rate constant k of 0.1164min-1could be obtained at pH 2.0, PW12concentration of 600mg/L and 4BE initial concentration of 50mg/L. The photocatalytic degradation rate of 4BE decreased with the increase of dye concentration when 4BE concentration was in the range of 50 to 150mg/L. On the basis of the results of cyclic voltammogram and the UV-vis spectrum of 4BE, the mechanism for the photodegradation of 4BE by phosphatotungstic acid includes three pathways: oxidation by hydroxyl radicals, electron transfer within the complex of the excited phosphatotungstic acid and 4BE, and reduction of dye by the reduced phosphatotungstic acid, in which the oxidation of hydroxyl radicals palys a leading role.
phosphatotungstic acid;homogeneous photocatalytic;direct red 4BE;reaction mechanism
X132
A
1000-6923(2011)06-0921-06
2010-10-16
国家自然科学基金资助项目(51009115);陕西省教育厅科研专助项目(07JK349);陕西省科技计划项目(2010JQ5008)
* 责任作者, 副教授, kebinli@sohu.com
魏 红(1977-),女,陕西大荔人,副教授,博士,主要从事有机污染防治与水资源保护方面的研究.发表论文近20篇.
猜你喜欢
化学工业与工程(2022年1期)2022-03-29
汽车工程师(2021年12期)2022-01-18
天津医科大学学报(2021年4期)2021-08-21
粮食与食品工业(2021年4期)2021-08-19
古今农业(2021年2期)2021-08-14
石油化工(2021年3期)2021-04-08
济宁医学院学报(2018年6期)2018-12-28
沈阳化工大学学报(2018年1期)2018-05-30
小溪流(画刊)(2017年3期)2017-03-23
印刷技术·数字印艺(2015年10期)2015-12-10
