
W形六角铁氧体BaFe18O27电子结构与导电性的第一性原理研究*
2011-11-02 03:26:38李忠虎李林朱林
物理学报 2011年10期
李忠虎李林 朱林
W形六角铁氧体BaFe18O27电子结构与导电性的第一性原理研究*
李忠虎1)2)李林1)朱林1)
1)(东北大学理学院物理系,沈阳110819)
2)(朝鲜理科大学物理系,平壤)
(2010年11月9日收到;2011年2月14日收到修改稿)
采用基于第一性原理的GGA+U方法研究了BaFe18O27的晶体结构和基态电子结构.以实验数据为初始结构的离子弛豫显示,由于稳定结构中离子半径的差异和2d位Fe的存在,位于BaO层中6h位的O离子脱离了实验结构中原胞的“表面”位置,产生畸变.计算得到晶体磁矩为28μB/f.u.,与实验相符.电子态密度及能带计算表明该材料具有微弱的半金属特性,而且与c轴平行方向和垂直方向的能带色散关系有着很大不同,6g位Fe在该材料的输运特性中起着关键作用,它们形成一种“导电层”,导致垂直电导率和平行电导率出现非常大的差异.
第一性原理,W形六角铁氧体,电子结构,导电性
PACS:71.15.Mb,75.50.Gg,72.80.Ga
1.引言
随着高科技的迅速发展,在我们的生活中不断出现高频段电子设备,电磁干扰与电磁辐射污染已成为引人注目的问题.为此,人们努力寻找一些能吸收微波段电磁辐射的材料(吸波材料).铁氧体磁材料因具有比较合适的导电性、介电性和磁性而呈现出较好的电磁波吸收性能,受到人们广泛关注[1—3].其中六角铁氧体具有较强的磁晶格向异性、良好的高频段特性和吸波性能.W形六角铁氧体与M形六角铁氧体相比其各向异性场较小,自然共振频率较低,更接近需要吸收的电磁波频段.BaFe18O27是W形六角铁氧体中最典型的母材料之一,能直接用作吸波材料或者调整其成分也能制备具有多样磁晶各向异性的吸波材料.六角铁氧体原胞中原子数目较多,第一性原理计算量较大,关于这类材料的电子结构与有关物理性质的计算研究基本限于M形六角铁氧体[4,5].最近Kníek等人[6]通过第一性原理研究了Y形六角铁氧体的电子结构及一些离子对导电性的影响.到目前为止尚未见到有关W形六角铁氧体的第一性原理研究的报道.一般实用的铁氧体大部分都是掺杂或者复合的材料.为了进一步研究掺杂型W形六角铁氧体,需要研究母材料BaFe18O27的电子结构以及导电来源.
本文通过基于第一性原理的广义梯度近似(GGA)计算方法研究了W形六角铁氧体的母材料BaFe18O27的基态电子结构与输运机理.
2.晶体结构及计算细节
2.1.晶体结构
按照化学成分和晶体结构,六角铁氧体可分成为M,W,X,Y,Z以及U形,如表1所示[2].表中Ba可替代为Sr,Me代表Fe2+,Ni2+,Co2+,Mn2+,Zn2+等二价金属阳离子,Fe全部为三价,S=Fe6O8(spinel,尖晶石型),R=BaFe6O11(六角),T= Ba2Fe8O14(六角),(*)号表示将相应块围绕六角轴旋转180°而得到的相应结构.实验研究发现BaW形六角晶体BaFe18O27是半导体,晶格常数为a =b=5.88,c=32.845,属于P63/mmc(194)点阵群[7].
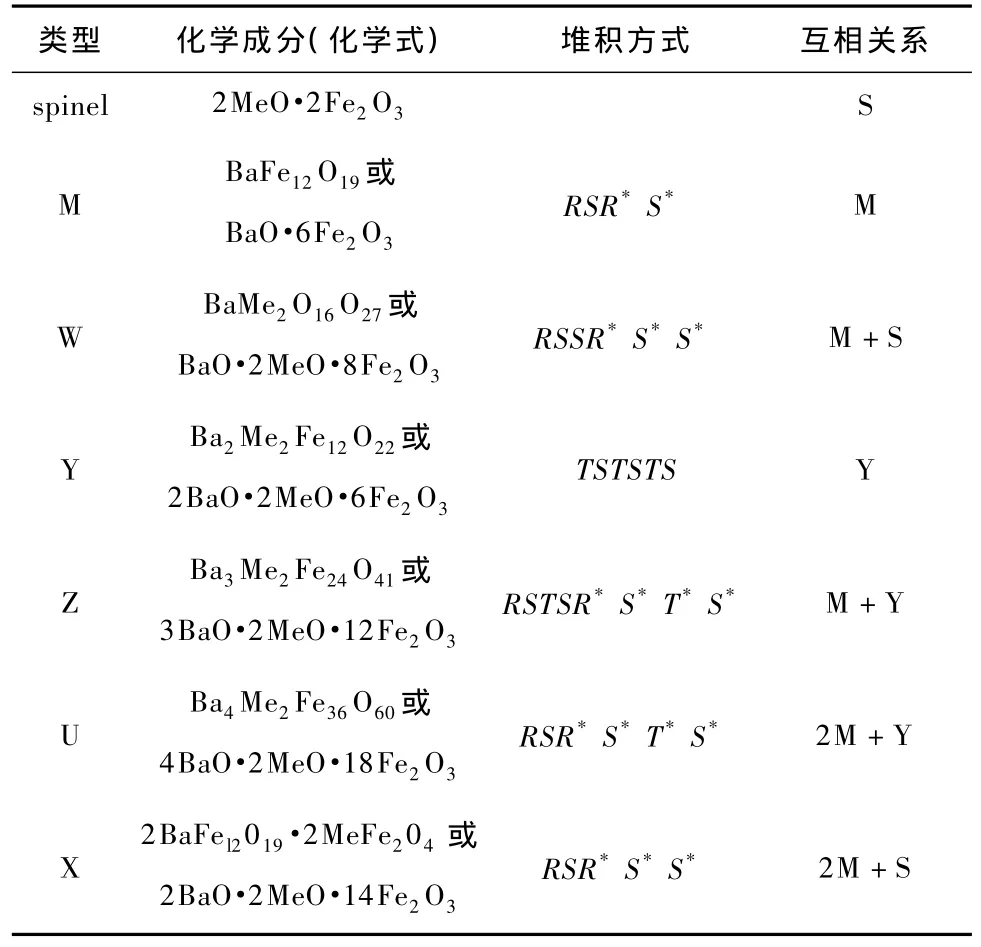
表1 不同类型六角铁氧体的化学成分与堆积结构
图1表示BaFe18O27晶体的原胞.这相当于两个单位化学式,是由2个Ba,36个Fe和54个O一共92个原子组成.

图1 BaFe18O27晶体原胞结构
W形铁氧体的金属离子的等效点位置(Wyckoff位置)和磁矩结构如表2所示[8].
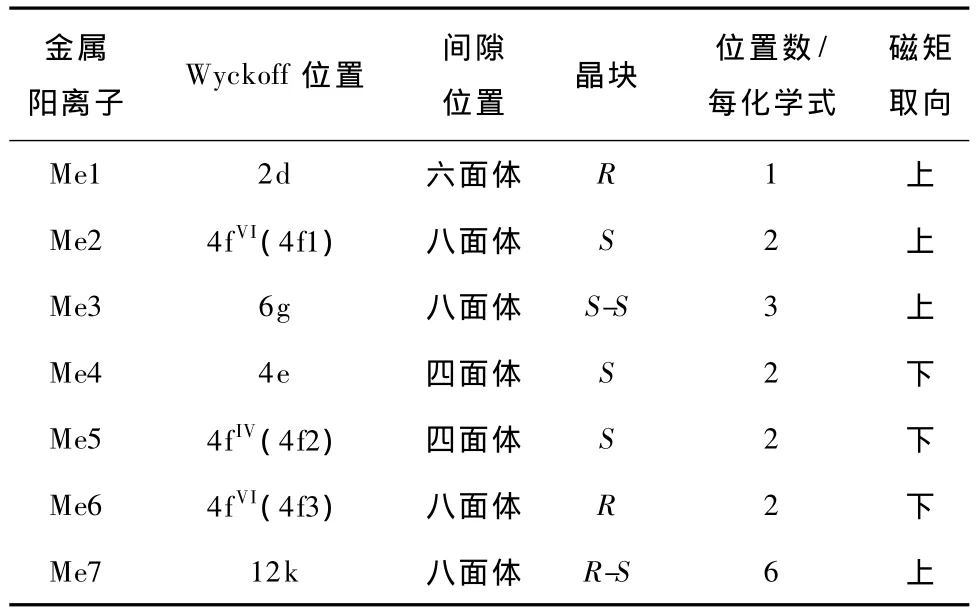
表2 原胞中金属离子的Wyckoff位置和磁矩取向
2.2.计算细节
计算采用了基于投影缀加平面波(PAW)方法的VASP程序包[9]以及PBE[10]形式的广义梯度(GGA)近似.Ba,Fe以及O的价带电子分别选取5 s25 p66 s2,3 d74 s1以及2 s22 p4.平面波动能截止能量设定为Ecut=500 eV.弛豫和静态计算时布里渊区积分的k点选用了以Gamma点为中心的8×8 ×2 Monckhorst-Pack网格,不可约布里渊区内有20个k点,这可以保证总能量误差不大于0.001 eV/原胞.再细的网格对总能量误差和优化结构的影响不大.为了得到稳定结构,以实验晶格常数[7]为初始值,每个Fe原子的初始磁矩取向按照Gorter模型[8]排列,对原胞进行了离子弛豫(包括原子坐标,原胞体积及形状),直到离子之间的Hellmann-Feynman力达到0.005 eV/以下为止.具体步骤是,先以实验值为初始值,在固定体积和形状之下用低精度(Ecut=400 eV,力低于0.2 eV/)优化结构,然后在这些优化结果基础上再用高精度(Ecut=50 0 eV,力低于0.0 05 eV/)进行全面弛豫而得到了稳定的晶体结构.总能量的电子自洽计算收敛判据设定为1 0-6eV.布里渊区积分使用了Gaussian展开方法,展宽能量选定为0.0 5 eV.
通常过渡金属氧化物因d电子局域性而呈现出强关联效应,用GGA方法不足以精确描述此类材料的电子结构.为了考虑Fe 3d电子对态密度的影响,我们还采用了GGA+U计算.在GGA+U计算中我们采用Dudarev[11]的有效参数Ueff=U-J,其中U和J分别为Hubbard和交换能参数,为简单起见文中用U代替Ueff.研究表明[4]Fe3O4和Fe2O3的八面体位和四面体位Fe的U值分别为7.33 eV—7.38 eV和6.33 eV,而对六角M形六角铁氧体BaFe12O19,U=6.94 eV是最符合于说明该材料的居里温度.本文采用U=7 eV来对Fe的3 d轨道进行GGA+U计算.
3.计算结果与分析
图2分别给出了结构优化前后得到的BaFe18O27晶体费米能级(Ef)附近的总态密度.下面所有的态密度图中上半部分为多数自旋电子的态密度而下半部分为少数自旋电子的态密度.

图2 BaFe18O27的总态密度(a)-GGA;(b)-GGA+U:优化前(实验晶格常数);(c)-GGA;(d)-GGA+U:优化后
从总态密度图上能看到基于实验晶格常数的GGA和GGA+U计算都得到金属态.实验表明此材料导电性属于半导体范围[7,12].GGA优化之后多数自旋电子态密度中出现能隙,GGA+U优化后无论多数自旋电子态密度还是少数自旋电子态密度都出现明显的能隙.比7 eV更大的U值计算只能增加能隙,不会对计算结果产生定性影响,费米能级一直位于少数自旋电子价带顶部,显示出微弱的半金属性.
GGA优化计算得到的晶格常数略大于实验值,由实验值的a=5.8800,c=32.845,c/a= 5.5859弛豫为a=5.8928,c=33.018,c/a= 5.8928.这是利用GGA-PBE赝势得到的晶格常数一般趋势[13].GGA+U优化计算得到的原胞中每个原子坐标与GGA计算结果基本相符,可晶格常数比GGA优化计算得到的稍大(U=7 eV时,a= 5.9372,c=33.159,c/a=5.5850).GGA+U优化得到的原胞中每个原子坐标的计算值如表3所示.
从表3上可以看到优化后原胞的Fe离子的位置基本上保持不变,但BaO层中6 h位的O离子脱离原胞“表面”位置(图3),位于菱形结构边心和中心的O离子从原位偏离.该变化一方面缘于O和Ba离子半径的差异(O2+:1.40,Ba+2:1.35),另外即便O和Ba离子半径相同,由于BaO层中三个氧离子组成的正三角中心位置上也存在着一个2d位的Fe离子,必然导致由Ba和O组成的正六角形的畸变.这种从理想结构偏离的现象在其他W形六角铁氧体的BaO层中也普遍存在[14].

图3 在Ba层中各离子的位置(a)弛豫前;(b)弛豫后

表3 BaFe18O27原胞的原子坐标
实验表明将具有离子半径与Fe2+(0.74)几乎相等的Co2+(0.72)掺杂到BaFe18O27时Co2+离子倾向于出现在S块-S块界面和S块的八面体6g和4 f1位上[13].6 g和4 f1位,尤其6 g位的周围环境类似于磁铁矿Fe3O4的B位置(八面体位).磁铁矿Fe3O4晶体是半金属,而且该材料的B位置一半被Fe2+占据而另一半被Fe3+占据[15].计算得到6 g位Fe的最近邻Fe—O间隔都在2.051—2.060范围之内.而在Fe3O4的B位置的最近邻Fe-O间隔为2.058,两者相差很小.
如果采用离子模型而把6 g位的Fe当中两个看做为二价(Fe2+,磁矩:4μB)并且其他位Fe离子都看做为三价(Fe3+,磁矩:5μB),则能得到原胞的每分子磁矩为28μB/f.u..优化后计算得到的原胞磁矩为56.00μB,即每分子磁矩为28.00μB/f.u.,与实验相符[8].
图4所示是每个等效点位置的Fe电子分态密度.由于Fe的s,p电子轨道的分态密度以及Ba和O的分态密度对费米能级附近态密度的贡献极小,图中只给出Fe 3 d电子的态密度分布.虽然图上没给出,但O离子当中对费米能级附近少数自旋电子态密度的贡献比较大的是6 g位Fe上下层的(4f2位和12k2位)O的2p电子.从图中可以看到对费米能级附近态密度的贡献最大的是6g位Fe 3 d电子.其他位置Fe的态密度对费米能级的贡献几乎为零.从与磁铁矿Fe3O4结构B位类似性可以推测6g位置之所以对费米能级附近态密度有如此大的贡献是因为在这个位置上大量的Fe2+和Fe3+混合在一起从而产生多余的导电电子.这类似于重掺杂的半导体-简并半导体的情况.
图5所示是多数自旋电子和少数自旋电子沿着高对称性k点的能带色散关系.图上看到自旋向上的多数自旋电子呈现半导体性质,而自旋向下的少数自旋电子呈现金属性质.能带图上沿着c轴的方向是G—A,H—K区域,其他的区域与c轴垂直方向.
从图上可以看到沿着c方向能带变化几乎为零,然而沿着与c轴垂直方向能带变化却很明显.在能带理论中电子的有效质量为m*=2/(2E(k) /k2).这里E(k)为布里渊区k点上的单电子本征能量(能带),为普朗克常数.虽然尚未报到与该材料的电子有效质量有关的研究资料,但是从图5可以看到沿着c轴平行方向的电子有效质量比垂直方向的大得多.
图6所示是由6g位Fe组成而与c轴垂直的平面内能量为费米能级附近(Ef±0.1 eV)的电子分布计算图.图中不难看出有些等高线已经不再是局域的而是分布在整个平面内.这表明6 g位Fe平面内已经有了自由载流子.
原胞中具有费米能级附近能量(Ef±0.1 eV)的电子分布如图7所示.
从图7中可以看到具有费米能级附近能量的电子大部分来自于6 g位Fe和它上下相邻的4 f2位O和12 k2位O.图6和图7说明它们形成一种“导电层”,以至此材料的导电基本上限于与c轴垂直的方向.输运理论中电导率为σ∝n/m*(其中n为载流子密度,m*为载流子有效质量).由图5—7可以推测与c轴垂直电导率比平行电导率高的多.实验研究发现垂直电导率比平行电导率高达103倍[12].因此,通过置换6g位的Fe离子或者改变它的电子性质可以极大程度改变该材料的输运特性.

图4 每个等效点位置的Fe 3 d电子分态密度

图5 沿着不同高对称性方向的能带色散(GGA+U,U=7 eV)(a)多数自旋电子;(b)少数自旋电子

图6 6g位Fe层中费米能级附近能量电子的对数分布图(单位:ρ·Vcell,其中ρ:电子密度,Vcell:原胞体积)

图7 原胞中具有费米能级附近能量的电子分布(图中等电子密度面的电子密度(ρ·Vcell)为0.01)
4.结论
密度泛函GGA+U方法研究了BaFe18O27的晶体结构与电子结构.计算结果表明,由于在BaO层中Ba和O的离子半径的差异以及2 d位Fe的存在,Ba和O不再形成理想的正六角形.另外,BaFe18O27呈现出微弱的半金属,磁矩为28μB,于实验相符.能带色散图上得知沿着c轴方向导电电子有效质量比与c轴垂直方向的大得多.S块的6 g位Fe处于与尖晶石晶体Fe3O4的八面体位Fe很相似的晶体环境而且费米能级附近少数自旋电子是基本上由它们的3 d电子组成.6 g位Fe和它们上下层O形成一种“导电层”,使垂直电导率比平行电导率高得多.
感谢复旦大学物理系车静光教授的建议和支持.
[1]Li Z W,Lin G Q,Wu Y P,Kong L B 2009 J.Phys.D 42 095007
[2]mitzgür,Yahya Alivov,Hadis Morko2009 J.Mater.Sci: Mater Electron 20 789
[3]Harris V G,Geiler A,Chen Y J,Yoon S D,Wu M Z,Yang A,Chen Z H,He P,Parimi P V,Zuo X,Patton C E,Abe M,Acher O,Vittoria C 2009 J.Magn.Magn.Mater.321 2035
[4]Novák P,Rusz J 2005 Phys.Rev.B 71 184433
[5]Fang C M,Kools F,Metselaar R,With G and Groot R A 2003 J.Phys.C 15 6229
[6]Kníek K,Novák P,Küpferling M 2006 Phys.Rev.B 73 153103
[7]Braun P B 1957 Philips.Res.Rep.12 491
[8]Gorter E W 1957 Proc.IEE 104 B(suppl.)255
[9]Kresse G,Furthmüller J 1996 Phys.Rev.B 54 11169
[10]Perdew J P,Burke K,Ernzerhof M 1996 Phys.Rev.Lett.77 3865
[11]Dudarev S L,Botton G A,Savrasov S Y,Humphreys C J,Sutton A P 1998 Phys.Rev.B 57 1505
[12]ima Z,Zalesskij,Závěta K 1966 Phys.Stat.Sol.14 485
[13]Mattsson A E,Armiento R,Schultz P A,Mattsson T R 2006 Phys.Rev.B 73 195123;Paier J,Marsman M 2006 J.Chem.Phys.124 154709
[14]Collomb A,Wolfers P,Obradors X 1986 J.Magn.Magn.Mater.62 57
[15]Zhou F,Ceder G 2010 Phys.Rev.B 81 205113
PACS:71.15.Mb,75.50.Gg,72.80.Ga
First-principles study of the electronic structure and electric conductivity in W-type hexagonalferrite BaFe18O27*
Ri Chung-Ho1)2)Li Lin1)Zhu Lin1)
1)(Department of Physics,Northeastern University,Shenyang 110819,China)
2)(Department of Physics,University of Science,Pyongyang,D.P.R.Korea)
(Received 9 November 2010;revised manuscript received 14 February 2011)
The electronic ground state and the electric conductivity of W-type hexagonal ferrite BaFe18O27are investigated in the generalized gradient approximation(GGA)as well as the GGA plus Hubbard U(GGA+U)scheme.The ionic relaxation calculation of the experimental crystal structure shows that oxygen ions at 6 h site in the BaO layer move away from the“surface”position in the unit cell,resulting in a structural distortion.The magnetic moment of the cell is calculated to be 28μB/f.u.,in agreement with previous experimental results.By taking account of electronic band structure and crystal ionic configuration it is found that the material is a weak half-metal and the effective mass of conduction electrons along the c axis is much heavier than that perpendicular to this axis.Fe ions on octahedral 6 g sites and O ions around them of the spinel block form a“conductive layer”.Therefore the electric conductivity perpendicular to the c axis is much greater than that parallel to the c axis.
first-principles,W-type hexagonal ferrite,electronic structure,electric conductivity
*辽宁省计划科学基金(批准号:2006222002)资助的课题.
E-mail:richungho@sohu.com
*Project supported by the Natural Science Foundation of Liaoning Province,China(grant No.2006222002).
E-mail:richungho@sohu.com
猜你喜欢
四川大学学报(自然科学版)(2025年1期)2025-02-10 00:00:00
小天使·聪聪画刊(2021年2期)2021-09-10 07:22:44
汽车零部件(2020年10期)2020-11-09 03:41:42
数学物理学报(2019年6期)2020-01-13 06:08:24
发明与创新·小学生(2019年12期)2019-12-05 06:02:28
表面工程与再制造(2019年3期)2019-09-18 01:35:10
汉语世界(The World of Chinese)(2019年6期)2019-09-10 07:22:44
电镀与环保(2017年3期)2017-06-23 08:24:52
深圳大学学报(理工版)(2015年5期)2015-02-28 16:21:26
无机化学学报(2014年12期)2014-02-28 17:34:04
