
仿刺参肠道多糖的纯化及理化分析
2011-10-28 03:22:04林威威王茂剑常耀光王共明
食品科学 2011年17期
林威威,张 健,王茂剑,*,常耀光,王共明
(1. 上海海洋大学食品学院,上海 200000;2. 山东省海洋水产研究所,山东 烟台 264006;3. 中国海洋大学食品科学与工程学院,山东 青岛 266003)
仿刺参肠道多糖的纯化及理化分析
林威威1,2,张 健2,王茂剑2,*,常耀光3,王共明1,2
(1. 上海海洋大学食品学院,上海 200000;2. 山东省海洋水产研究所,山东 烟台 264006;3. 中国海洋大学食品科学与工程学院,山东 青岛 266003)
目的:对仿刺参肠道多糖进行纯化,并对纯化产物进行理化分析。方法:使用葡聚糖凝胶(G-100)层析柱分离出两种仿刺参肠道多糖,选取其中多糖A进行研究。使用葡聚糖凝胶(G-100)柱层析法和琼脂糖凝胶电泳法进行纯度检测,葡聚糖凝胶(G-200)柱层析测定分子质量,高效液相色谱法确定其单糖组分,并结合红外吸收光谱法与核磁共振对其结构进行初步检测。结果:实验所得纯化产物多糖A为均一组分,主要含有氨基糖醛酸、氨基半乳糖盐酸盐和岩藻糖3种单糖,并含有少量的氨基葡萄糖、半乳糖。其中岩藻糖有α和β两种构型,以α-岩藻糖硫酸酯残基、β-岩藻糖硫酸酯残基形式存在。分子质量约为18.8×104D。结论:该纯化产物多糖A为一种酸性多糖,推测其为硫酸软骨素类。
肠道多糖;纯化;单糖分析;结构鉴定;仿刺参
多糖是存在于自然界的醛糖和酮糖通过糖苷键连接在一起的多聚物,广泛存在于各种生物体中,是生命活动的基础物质之一,在生命活动中发挥着重要的作用。研究显示[1],多糖具有抗肿瘤活性、抗病毒、降血糖、抗炎、抗补体、抗辐射及解毒等生物活性。海参多糖是海参体内最重要的活性物质之一,存在于海参体内的多糖主要分为两类,一类为海参糖胺聚糖或黏多糖,另一类为海参岩藻多糖。海参多糖的提取纯化技术已较为成熟,但研究对象大多针对海参体壁。本实验基于对海参体壁多糖研究的较为成熟的技术,对仿刺参肠道多糖进行提取纯化研究。主要针对纯化技术进行参数确定,制备出仿刺参肠道纯化多糖,对其进行成分鉴定及结构分析,为进一步的功能性质鉴定提供实验依据。
1 材料与方法
1.1 材料与试剂
鲜仿刺参肠,产于山东省烟台市。
葡聚糖凝胶(Sephadex G-100)、葡聚糖凝胶(Sephadex G-200);葡聚糖标准品(Dextran Standard 1.2×104、5 × 104、15 × 104、27 × 104D);D-甘露糖 (Man)、D-(+)-氨基葡萄糖(GlcN)、D-氨基糖醛酸(GlcUA)、D-(+)-半乳糖醛酸(GalUA)、乳糖(Lac)、D-(+)-氨基半乳糖盐酸盐 (GalN)、D-半乳糖 (Gal)、L-阿拉伯糖(Ara)、L-岩藻糖 (Fuc)标准品 美国Sigma公司;其他试剂均为国产分析纯。
1.2 仪器与设备
Aglient1100型高效液相色谱仪(配有ZORBAX Eclipse XDB-C18分离柱4.6mm×150mm,5μm) 美国Agilent公司;Bruker AVANCE III 600 超导高分辨核磁共振谱仪、Bruker TENSOR 27傅里叶红外光谱仪 德国Bruker光公司;TU-1810PC紫外-可见分光光度计 北京普析通用仪器有限公司;Hel-VAP旋转蒸发仪 德国Heidolph公司;FDU-1200冷冻干燥机 日本Eyela公司;DYY-12C型电泳仪 北京市六一仪器厂。
1.3 方法
1.3.1 仿刺参肠道多糖分离纯化
取新鲜参肠,洗净后用筛沥水至无水滴下,投入组织捣碎机捣碎,匀浆。使用木瓜蛋白酶解法[2]提取,加酶量10400U/g,55℃条件下酶解6h,90℃条件下灭酶15min。将粗多糖溶液进行减压蒸发浓缩,取浓缩液加入1/2体积50% 的三氯乙酸(TCA)溶液,静置过夜。再于15000r/min条件下离心15min,取上清液加入醋酸钾使其浓度达到1.5mol/L,于4℃条件下静置过夜,15000r/min离心15min,收集沉淀,无水乙醇洗涤,得精制肠道多糖。
精制后肠道多糖使用柱层析法[3-5]进行纯化。肠道精制多糖用蒸馏水复溶成1.5mg/mL水溶液,取2mL上样至Sephadex G-100层析柱中,去离子水洗脱,恒定流速0.25mL/min,自动部分收集器收集,每管2mL,共收集80管。苯酚硫酸法[6-7]检测所收集溶液吸光度,按不同出峰分别采收,冷冻干燥得海参肠道纯化多糖。
1.3.2 纯度测定
采用凝胶柱层析与琼脂糖凝胶电泳法同时检测,凝胶柱层析法如1.3.1节中所述。
琼脂糖凝胶电泳[8]分析:缓冲液:0.06mol/L pH8.6巴比妥缓冲液;固定剂:0.1g/100mL十六烷基三甲基溴化铵溶液;染色液:以脱色液为溶剂配制0.1g/100mL甲苯胺蓝溶液;脱色液:V乙酸:V乙醇:V水=0.1:5:5;电泳条件:200mA条件下电泳60min。
称取0.24g琼脂糖加入30mL巴比妥缓冲液中,加热制胶。将纯化样品配制成4mg/mL的溶液,取两平行(一加溴酚蓝示踪剂,一为单样物质),上样8μ L。电泳结束后固定剂中固定1h,蒸馏水冲洗胶体表面,冲洗后置于甲苯胺蓝溶液中染色10min,再以脱色液漂至背景无色。
1.3.3 分子质量测定
葡聚糖标准曲线制作:将分子质量分别为1.2×104、5×104、15×104、27×104D 的葡聚糖标准品配制成1mg/mL水溶液,使用Sephadex G-200色谱柱依次上柱,上样量为1mL,去离子水洗脱,流速0.35mL/min,自动收集器收集,苯酚硫酸法检测,分别得各自洗脱体积Ve,27×104D洗脱体积作为外水体积Vo,以Ve/Vo为纵坐标,lgM为横坐标,绘制标准曲线[9-10]。
取纯化多糖以相同条件过柱,并收集计算洗脱体积,通过对比标准曲线计算分子质量。
1.3.4 单糖组成测定
采用柱前衍生高效液相色谱法[11-13]。
单糖标准品衍生物的制备:将M a n、G l c N、GlcUA、GalUA、GalN、Gal、Ara、Fuc 8种单糖标准品和内标物Lac配制成约0.36g/L的溶液,分别吸取50μL加入试管中,然后加入450μL 0.5mol/L的1-苯基-3-甲基-5-吡唑啉酮(PMP)甲醇溶液和等体积的0.3mol/L NaOH溶液,漩涡混匀,于70℃水浴中反应30min,取出冷却至室温,用450μL 0.3mol/L HCl溶液中和,加入1mL氯仿萃取,充分振荡,吸弃下层有机相,重复3次。将上层水相过膜,取1 0μL进样。
样品制备[14]:称取纯化样品2.0mg于安培瓶中,加入1mL 2mol/L 三氟乙酸(TFA),充氮气封管,于110℃水解8h。冷却至室温,5 0℃挥干TFA,逐次以2、0.3mol/L NaOH溶液缓慢调至中性,定容至1mL,取其中400 μ L并混入50 μ L内标物Lac进行PMP衍生化。具体方法同上所述。
色谱条件:Agilent 1100高效液相色谱仪,搭配ZORBAX Eclipse XDB-C18分离柱(4.6mm×150mm,5 μ m),紫外检测器(254nm);流速:1.0mL/min;柱温:25℃;流动相A:15%乙腈+0.05mol/L磷酸缓冲液(KH2PO4-NaOH,pH6.9);流动相B:40%乙腈+0.05mol/L磷酸缓冲液(KH2PO4-NaOH,pH6.9);时间梯度:0→10→30min;浓度梯度:0→8%→20%流动相B;进样体积:10μ L。
1.3.5 波谱测定
红外光谱:采用KBr压片法进行红外光谱扫描,取纯化样品1mg,与200mg KBr混合研磨,压片后,再以傅里叶变换红外光谱仪进行扫描,扫描波数范围为4000~400cm-1。
核磁共振(NMR):称取样品50mg,0.5mL D2O溶解后,场强600MHz,上机分析。
2 结果与分析
2.1 仿刺参肠道组织精制多糖柱层析
仿刺参肠道组织精制多糖经Sephadex G-100洗脱后,根据苯酚硫酸法检测,以收集管号为横坐标,吸光度为纵坐标作图。
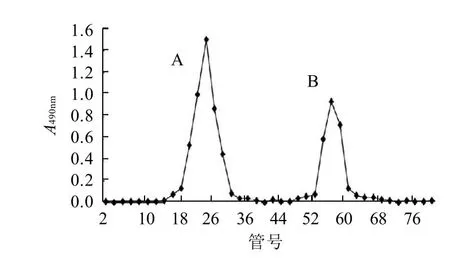
图1 仿刺参肠道组织精制多糖Sephadex G-100柱层析洗脱曲线Fig.1 Elution curve of purified polysaccharides from intestinal tract of Apostichopus japonicus
如图1所示,仿刺参肠道精制多糖经过柱层析后,产生两个显著峰A和B,两峰无重叠无拖尾,分离效果较好。本实验以峰A为研究对象进行分部收集,经冷冻干燥后得到纯化多糖A,称质量后计算其提取率为0.4‰(B得率相对较少,目前仍在收集过程中)。
2.2 纯度测定结果
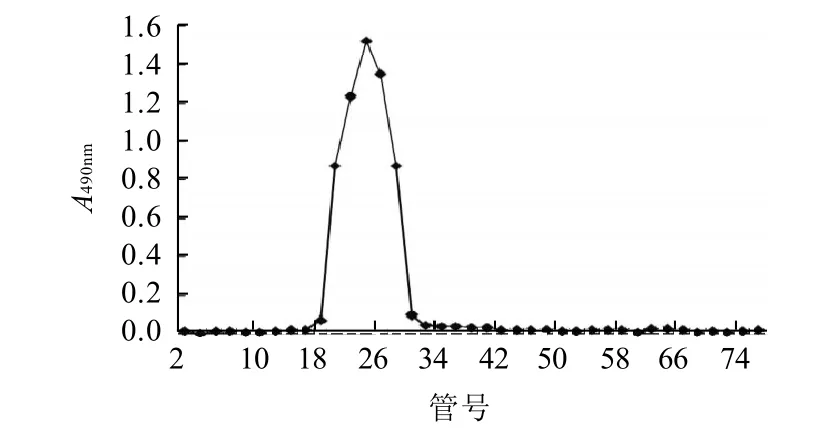
图2 仿刺参肠道组织纯化多糖Sephadex G-100柱层析图Fig.2 Gel column chromatography curve of polysaccharide A
如图2所示,精制多糖经过层析柱纯化后出峰单一集中,说明分子质量大小属于同一范围的多糖被集中洗脱出来。

图3 仿刺参肠道纯化多糖琼脂糖凝胶电泳图Fig. 3 Agarose gel electrophoresis of polysaccharide A
如图3所示,第5与第8泳道上两点均为多糖A的琼脂糖凝胶电泳扫描,其中第5泳道上点为添加溴酚蓝示踪剂的平行样,第8泳道上点为多糖A单物质。两点均清晰无拖尾,说明多糖A为均一成分,纯度可达到电荷均一。
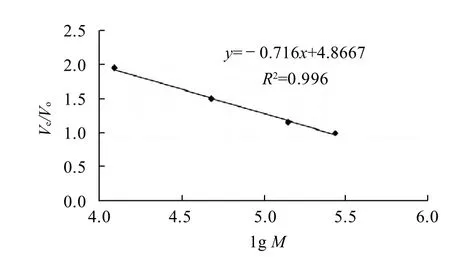
图4 葡聚糖分子质量标准曲线Fig. 4 Calibration curve for relative molecular weight determination
2.3 分子质量测定结果根据图4标准曲线计算,海参肠道纯化多糖A的分子质量约为18.8×104D。
2.4 单糖组分测定结果
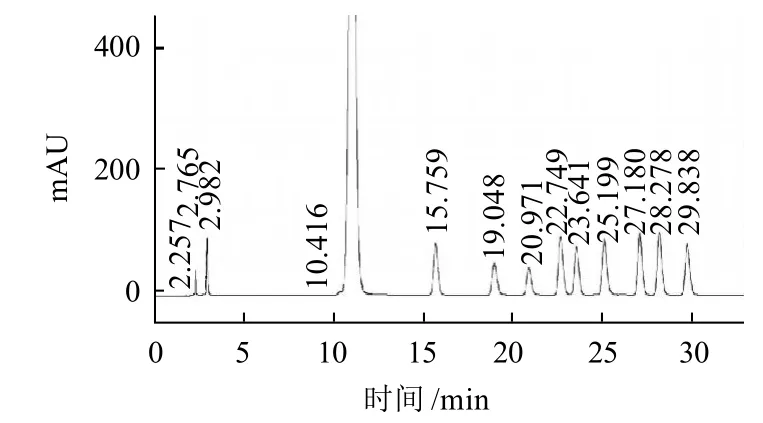
图5 单糖标准品高效液相色谱图Fig. 5 HPLC of standard monosaccharides
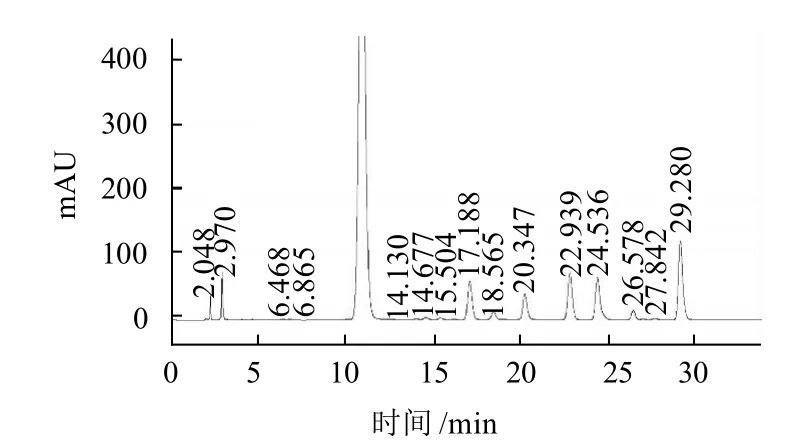
图6 样品单糖组分高效液相色谱图Fig. 6 HPLC of monosaccharides in polysaccharides A
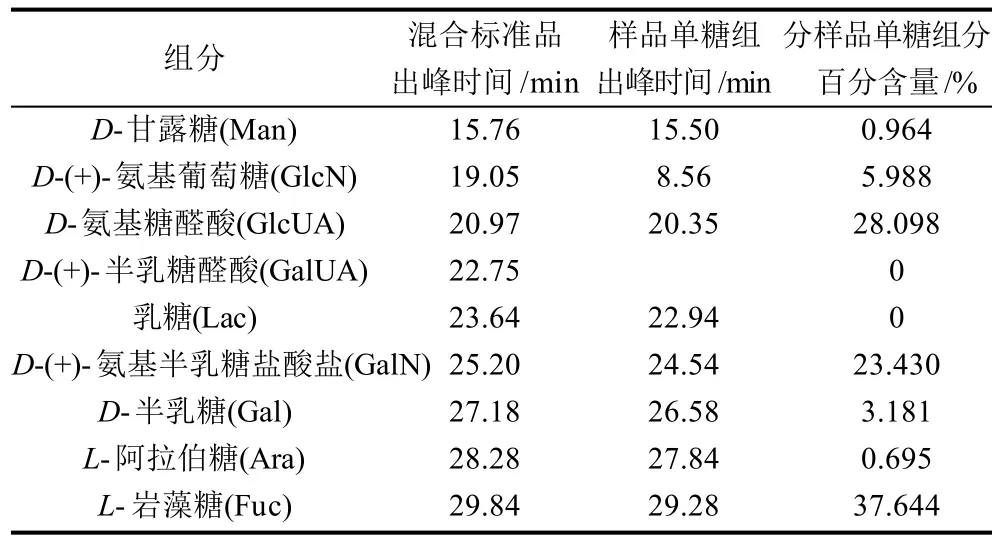
表1 仿刺参肠道纯化多糖组分分析Table 1 Monosaccharide composition analysis of polysaccharides A
结果如图5、6及表1所示。仿刺参肠道纯化多糖A主要由氨基糖醛酸、氨基半乳糖盐酸盐和岩藻糖等3种单糖组成,其含量分别为28.098%、23.430%、37.644%,并含有少量的氨基葡萄糖(5.988%)、半乳糖(3.181%),微量的甘露糖和阿拉伯糖。多糖A的单糖组分与构成海参硫酸软骨素的单糖种类相符[15],因此可推测多糖A为硫酸软骨素类多糖。
2.5 红外图谱分析

图7 纯化产物红外吸收光谱Fig. 7 Infrared absorption spectrum of polysaccharides A
根据参考文献[16-19]分析,3447.74cm-1为-OH特征吸收峰,表明存在分子间、分子内氢键,2925.22cm-1处为C—H的伸缩振动,此两组峰为多糖的特征峰;2000~1500cm-1是双键伸缩振动区,其中1636.34cm-1可能是C=O的伸缩振动峰;1500~1300cm-1区域主要提供了C-H弯曲振动的信息;1250.11cm-1是S=O的伸缩振动,818.88cm-1处出现C-O-S拉伸振动吸收峰,表明该糖含有硫酸酯基;1200~1000cm-1为吡喃糖环的C-O伸缩振动所引起的,其中1165.91cm-1处为C-O-H的振动峰,1029.70cm-1归属于糖环上C-O-C的伸缩振动;963.43、928.98cm-1显示了环的骨架振动;902.61cm-1处可能是β-型吡喃糖环C-H变角振动引起的。因此认为多糖A可能为一种含硫酸酯基的β-吡喃多糖。
2.6 核磁共振分析
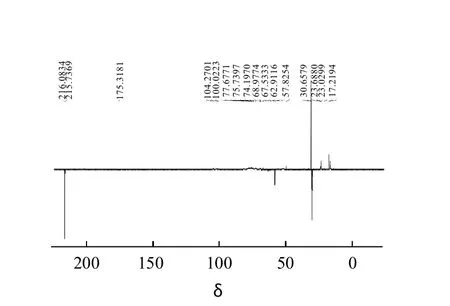
图8 纯化产物13C核磁共振图Fig. 8 13C nuclear magnetic resonance spectrum polysaccharides A
结合参考文献[18-22],根据13C的DEPT-135°图谱(图8),57 ×10-6~104×10-6范围的碳信号表明吡喃糖的存在,δ=100以上糖信号显示着半乳糖或岩藻糖的存在,δ=16附近信号的存在证实α-岩藻糖残基的存在,61×10-6~81 ×10-6的信号是岩藻聚糖C2、C3、C4的信号峰,并且存在着取代基,如硫酸酯键或糖苷键。20×10-6~57×10-6是β-岩藻糖残基上的C6的CH3信号峰,另外,岩藻糖的α构型和β构型在δ=100.0223、δ=101.2482、δ=104.2701也得到了体现。
δ=57.8254处的负峰则对应-CH2OH基团,推测分子内存在半乳糖残基,δ=175.3181表明了多糖A中含有-COOH。从DEPT-135°碳谱进一步推测分子内存在半乳糖残基和半乳糖醛酸残基。

图9 纯化产物1H核磁共振图Fig. 9 1H nuclear magnetic resonance spectrum of polysaccharides A
依据氢谱(图9),1.1×10-6~1.2 ×10-6和3.5×10-6~5.5×10-6信号能归属于α-岩藻糖残基,相对应的δ=1.9附近的信号峰和3.5×10-6~ 5.5×10-6的信号能归属于β-岩藻糖残基,δ=1.0附近信号应为-CH3的质子峰。该结果和碳谱的信号峰相一致。
结合红外光谱、碳谱以及氢谱的信号综合分析,该分子内存在α-岩藻糖硫酸酯残基、β-岩藻糖硫酸酯残基、半乳糖残基和半乳糖醛酸残基。
3 结 论
通过实验发现,葡聚糖G-100对纯化仿刺参肠道多糖效果良好,能够分出两个不重叠无拖尾的多糖峰,其中纯化多糖A的提取率为0.4‰,纯度检测结果证实多糖A分子质量集中分布,具有电荷均一性。对肠道多糖A成分进行了单糖组分分析,其中主要含有氨基糖醛酸、氨基半乳糖盐酸盐和岩藻糖3种单糖,还有少量的氨基葡萄糖、半乳糖,微量的甘露糖和阿拉伯糖。波谱分析结果与高效液相所检测单糖组分相吻合,且可知多糖A中岩藻糖存在α和β两种构型,以α-岩藻糖硫酸酯残基、β-岩藻糖硫酸酯残基形式存在,为一种酸性黏多糖,推测属于硫酸软骨素类。肠道多糖中另一分子质量较小的B成分仍需继续研究。
[1] 方积年, 丁侃. 天然药物: 多糖的主要生物活性及分离纯化方法[J].中国天然药物, 2007, 5(5): 388-345.
[2] 陈涛, 张健, 王茂剑. 木瓜蛋白酶提取仿刺参消化道多糖的研究[J].食品科学, 2010, 31(20): 226-229.
[3] 迟玉森, 仇宏伟, 庄桂东, 等. 长岛刺参多糖得提取精制及基本性质[J]. 精细化工, 2007, 24(5): 480-483.
[4] 蔡彬新, 吴成业. 海参多糖的分离纯化方法及其主要生物活性[J]. 福建水产, 2008, 9(3): 70-74.
[5] 田亚平. 生化分离技术[M]. 北京: 化学工业出版社, 2006: 69-97.
[6] 严成, 严夏. 枸杞多糖提取工艺比较及体外抗氧化性研究[J]. 食品科学, 2008, 29(7): 183-187.
[7] 钟建平, 钟春燕, 赵道辉, 等. 苯酚硫酸比色法测定保健食品多糖的研究[J]. 中国卫生检验杂志, 2001, 11(6): 675-675.
[8] 张惟杰. 糖复合物生化研究技术[M]. 2版. 浙江: 浙江大学出版社,1999: 401.
[9] YANG Jianhong, DU Yumin, HUANG Ronghua, et a1. Chemical modification and antitumour activity of Chinese lacquer polysaccharide from lac treeRhus vernicifera[J]. Carbohydrate Polymers, 2005,59: 101-107.
[10] 徐琴, 徐增莱 , 沈振国 , 等. 山药多糖的研究[J]. 中药材, 2006, 29(9): 909-912.
[11] 戴军, 朱松, 汤坚, 等. PMP柱前衍生高效液相色谱法分析杜氏盐藻多糖的单糖组成[J]. 分析测试学报, 2007, 26(2): 206-210.
[12] YOO S H, YOON E J, CHA J, et a1. Antitumor activity of levan polysaccharides from selected microorganisms[J]. Int J Biol Macromol,2004, 34(1/2): 37-41.
[13] FU D T, ONEILL R A. Monosaccharide composition analysis of oligosaccharides and glycoproteins by high-performance LIQUID chromatography[J]. Analytical Biochemistry, 1995, 227(2): 377-384.
[14] YUTAKA K, SHUGO W, MAMORU K, et al. Structure of fucose branches in the glycosaminoglycan from the body wall of the sea cucumberStichopus japonicus[J]. Carbohydrate Res, 1997, 297(39):273-279.
[15] 尹利昂, 陈士国, 薛长湖, 等. 4种海参中含岩藻糖支链的硫酸软骨素化学组成差异的分析[J]. 中国海洋大学学报, 2009, 39: 63-68.
[16] 夏朝红, 戴奇, 房韦, 等. 几种多糖的红外光谱研究[J]. 武汉理工大学学报, 2007, 29(1): 45-47.
[17] 尹利昂. 不同海参多糖的分离纯化及生化性质分析[D]. 青岛: 中国海洋大学, 2009.
[18] 李熙灿, 曾和平, 郑雨, 等. 光谱法鉴定黑海参中的多糖成分[J]. 中国药房, 2005, 16(13): 1012-1014.
[19] 葛淑敏, 于源华, 张艳飞. 蒙古口蘑多糖组分分析及结构初步鉴定[J]. 安徽农业科学, 2009, 37(1): 192-193.
[20] YAMADA K, MATSUBARA R, KANEKO M, et a1. Constituents of holothuroidea.10.1)isolation and structure of a biologically active ganglioside molecular species from the sea cucumberHolothurialeucospilota[J]. Pharmaceutical Society of Japan, 2001, 49(4): 447-452.
[21] VIEIRA R P, MULLOY B, MOURAO P A. Structure of a fucosebranched chondroitin sulfate from sea cucumber[J]. The Journal of Biological Chemistry, 1991, 266(25): 13530-13536.
[22] 刘密新, 罗国安, 张新荣, 等. 仪器分析[M]. 2版. 北京: 清华大学出版社, 2009: 170-211.
Purification and Physicochemical Analysis of Polysaccharides from Intestinal Tract ofApostichopus japonicus
LIN Wei-wei1,2, ZHANG Jian2, WANG Mao-jian2,*,CHANG Yao-guang3,WANG Gong-ming1,2
(1. College of Food Science & Technology, Shanghai Ocean University, Shanghai 200000, China;2. Marine Fisheries Research Institute of Shandong Province, Yantai 264006, China;3. College of Food Science and Engineering, Ocean University of China, Qingdao 266003, China)
Objective: To purify and physicochemically analyze polysaccharides from the intestinal tract ofApostichopus japonicus. Methods: Two polysaccharides, named as polysaccharides A and B, respectively, were isolated and purified by using SephadexG-100 column from the intestinal tract ofApostichopus japonicus, and polysaccharide A was selected to study its physicochemical properties. Its purity was evaluated by SephadexG-100 column chromatography and agarose gel electrophoresis.The relative molecular mass was determined by Sephadex G-200 column chromatography. Monosaccharide components were analyzed by high performance liquid chromatography (HPLC) and their structures were identifiedby infrared spectroscopy and nuclear magnetic resonance. Results: The purified polysaccharide A was composed of GlcUA, GalN, Fuc, and a small amount of GlcN and Gal. Fucose had two configurations including the forms ofα-fucose sulfate andβ-fucose sulfate residues. This polysaccharide had a relative molecular mass of approximately 18.8 × 104D. Conclusion: The purified polysaccharide A is an acidic polysaccharide, suggesting that it belongs to the chondroitin sulfate class.
intestinal tract polysaccharide;purification; component analysis;structure identification;Apostichopus japonicus
TS254.1
A
1002-6630(2011)17-0118-05
2011-06-30
国家海洋局海洋公益性行业科研专项(201105029);国家海洋局海洋经济规划科技推进平台与运作项目(国海科字[2007]219)
林威威(1985—),女,硕士研究生,研究方向为食品科学与工程。E-mail:742239054@qq.com
*通信作者:王茂剑(1964—),男,研究员,本科,研究方向为食品科学与工程。E-mail:wangmaojian@126.com
猜你喜欢
红蜻蜓·低年级(2021年12期)2022-01-19 05:18:32
红蜻蜓·低年级(2021年12期)2021-12-19 15:06:23
大连海洋大学学报(2020年2期)2020-05-06 02:26:10
吉林农业(2019年6期)2019-06-11 03:10:30
天然产物研究与开发(2019年1期)2019-03-01 05:41:20
中成药(2017年10期)2017-11-16 00:50:15
中成药(2017年4期)2017-05-17 06:09:46
天然产物研究与开发(2016年1期)2016-06-05 10:29:25
中南民族大学学报(自然科学版)(2015年2期)2015-12-16 12:11:10
食品科学(2013年24期)2013-03-11 18:30:30
