
GC 测定细辛药材中的农药残留量
2011-08-07 02:22王希尧田春莲鄢景森贺凤伟赵春杰辽宁科技学院本溪市70沈阳药科大学药学院沈阳市006
中国药房 2011年31期
王希尧,田春莲,鄢景森,贺凤伟,赵春杰#(.辽宁科技学院,本溪市70;.沈阳药科大学药学院,沈阳市 006)
中药材种植过程中使用的农药种类多达几百种,包括有机氯、有机磷、拟除虫菊酯氨基甲酸盐等多种类型[1,2],农药残留是因农药使用而残留在人类或动、植物体中的混合物[3]。农药进入药材及土壤中,经过食物链可以危害人的健康。有机氯类农药价廉、杀虫效率高,曾广泛被应用,造成环境污染;且其具有毒性,可造成慢性中毒或致畸、致癌、致突变等,还易在动植物体内蓄积,因而世界各国已相继禁止使用该类农药[4]。因此,笔者采用超声提取和弗罗里硅土柱净化,对细辛药材进行前处理,采用毛细管气相色谱-电子捕获检测器(GC-ECD)法对不同产地的细辛药材中的10种农药残留进行同时测定,结果个别样本有一定量的农药残留被检出。本方法简单,能广泛应用于农药残留的测定,规范药材的质量。
1 仪器与试药
Agilent 6890型气相色谱仪、Agilent 63Ni电子捕获检测器(美国Agilent公司);OV-1701弹性石英毛细管柱(大连中汇达公司)。
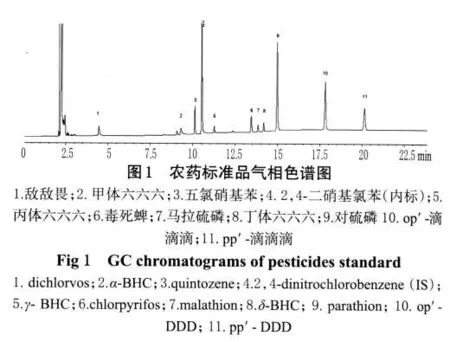
农药标准品:敌敌畏(Dichlorvos)、六六六(benzenehexachloride,BHC)[包括甲体六六六(α-BHC)、丙体六六六(γ-BHC)和丁体六六六(δ-BHC)3种异构体,纯度>99%]、五氯硝基苯(Quintozene)、毒死蜱(Chlorpyrifos)、对硫磷(parathion)、马拉硫磷(malathion)、2,4-二硝基氯苯(CDNB)均购自国家农药标准品中心;滴滴涕(DDT)包括op′-滴滴滴(op′-DDD)和 pp′-滴滴滴(pp′-DDD)2种同系物,纯度>98%,均购自美国Sigma公司;正己烷为色谱纯,水为重蒸馏水,其他试剂均为分析纯;所有药材样品(来源见表1)经沈阳药科大学中药学院贾景明副教授鉴定均为北细辛Asarum heterotropoidesFr.Schmidt var.mandshuricum(Maxim.)Kitag.。
2 方法与结果
2.1 色谱条件
色谱柱:OV-1701弹性石英毛细管柱(30 m×0.32 mm×0.5 μm);柱升温程序:初始温度 180℃,以 7℃·min-1升至260℃保持 2 min,以15℃·min-1升至 270℃保持10 min;进样口温度:260℃;检测器温度:300℃;载气:高纯氮,99.999%;流量:60 mL·min-1;进样方式:不分流进样;进样量:1 μL。
2.2 标准曲线的制备
精密称取敌敌畏、甲体六六六、五氯硝基苯、丙体六六六、毒死蜱、马拉硫磷、丁体六六六、对硫磷、op′-滴滴滴、pp′-滴滴滴各2.5 mg,分别置于25 mL量瓶中,加正己烷溶解,定容,于4℃贮藏,备用。
分别精密吸取敌敌畏、甲体六六六、五氯硝基苯、丙体六六六、毒死蜱、马拉硫磷、丁体六六六、对硫磷、op′-滴滴滴、pp′-滴滴滴贮备液适量,置于50 mL量瓶中,加正己烷稀释至刻度,制成每1 mL含敌敌畏 4.946 μg、甲体六六六2.32 μg、五氯硝基苯1.11 μg、丙体六六六45.2 μg、毒死蜱 120.0 μg、马拉硫磷 8.38 μg、丁体六六六2.61 μg、对硫磷 4.092 μg、op′-滴滴滴 1.13 μg、pp′-滴滴滴 1.15 μg的混合对照品贮备液。分别精密吸取上述混合对照品贮备液0.005、0.01、0.05、0.1、0.2、0.5、1 mL于10 mL量瓶中,用正己烷稀释至刻度,制备成7个浓度的混合对照品溶液,加入等体积的2,4-二硝基氯苯溶液使各溶液浓度均为4.4 μg·mL-1,常温下氮气流吹干至刻度。
分别精密吸取上述7份标准溶液各1 μL,按“2.1”项下色谱条件进样,每份进样5次,以样品峰面积与2,4-二硝基氯苯峰面积的比值(Y)为纵坐标,样品浓度(X)为横坐标,进行线性回归,回归方程见表2;气相色谱见图1。

表1 细辛药材样品来源Tab 1 Origin of Asari Radix et Rhizoma medicinal materials

表2 回归方程(n=5)Tab 2 Regression equation(n=5)
结果表明,敌敌畏在1.978×102~3.957×104ng·mL-1浓度范围内,甲体六六六在4.64~9.28×102ng·mL-1浓度浓度范围内,五氯硝基苯在1.554×102~3.108 ×104ng·mL-1浓度范围内,丙体六六六在4.52~9.04×102ng·mL-1浓度范围内,毒死蜱在2.74~5.472×102ng·mL-1浓度范围内,马拉硫磷在4.19×10~8.380×103ng·mL-1浓度范围内,丁体六六六在 5.22~1.044×103ng·mL-1浓度范围内,对硫磷在8.184×10~1.637×104ng·mL-1浓度范围内,op′-滴滴滴在 4.972×10~9.944 ×103ng·mL-1浓度范围内,pp′-滴滴滴在5.06×10~1.012×104ng·mL-1浓度范围内与各自峰面积与2,4-二硝基氯苯峰面积比值呈良好线性关系。各农药回归方程的相关系数为0.990 3~0.999 1,仪器精密度为1.2%~3.4%,方法精密度为6.5%~7.4%,加样回收率为84.0%~103.2%,RSD为2.5%~8.8%。
2.3 供试品溶液的制备
取药材约2 g,精密称定,置于50 mL锥形瓶中,加丙酮-水(7∶3)混合溶液40 mL,摇匀,超声处理15 min,抽滤,药渣再重复上述操作1次后,合并滤液。滤液转入250 mL分液漏斗中,加20 mL 5%NaCl溶液,振摇萃取,无机相再分别用60 mL正己烷萃取2次,合并正己烷相于250 mL磨口瓶中,于50~60℃水浴上旋转蒸发至约2 mL,作为提取液。通过20 mL正己烷预淋的层析柱(层析柱由下至上:少许玻璃棉+2 g无水硫酸钠+5 g弗罗里硅土+2 g氧化铝+2 g无水硫酸钠),再用50 mL丙酮-正己烷(1∶4)溶液进行洗脱,洗脱液收集于250 mL磨口烧瓶中,旋转蒸发至约2 mL,再转入10 mL容量瓶,用正己烷定容,即得。
2.4 出峰顺序
照“2.1”项下色谱条件进样测定。结果,出峰顺序为:敌敌畏、甲体六六六、五氯硝基苯、丙体六六六、毒死蜱、马拉硫磷、丁体六六六、对硫磷、op′-滴滴滴、pp′-滴滴滴。
2.5 色谱系统精密度试验
精密吸取同一浓度对照品溶液1 μL,重复进样5次,按“2.1”项下色谱条件测定。结果,敌敌畏、甲体六六六、五氯硝基苯、丙体六六六、毒死蜱、马拉硫磷、丁体六六六、对硫磷、op′-滴滴滴、pp′-滴滴滴的 RSD分别为 3.0%、2.4%、2.8%、1.2%、3.0%、2.6%、2.2%、2.4%、2.1%、3.4%。
2.6 方法精密度试验
精密称取1号样品2 g,共5份,放置12 h,按供试品制备方法制备供试品溶液,按“2.1”项下色谱条件进样测定。结果,丙体六六六、对硫磷、op′-滴滴滴的RSD分别为6.7%、7.4%和6.5%。
2.7 回收率试验
精密称取1号样品2 g,共9份,分3个水平添加混合标准品溶液,每个水平重复3次,混匀。按上述条件提取、净化、测定并计算回收率,结果见表3。
2.8 供试品的测定
精密称取各药材2 g,按供试品提取和净化方法操作项下平行制备样品各3份,按“2.1”项下色谱条件测定。每份样品重复2次,每次进样1 μL,用内标法定量。样品中农药残留量检测结果见表4(表中“ND”为低于检测线)。
3 讨论
笔者采用超声提取和弗罗里硅土柱净化,对样品进行前处理,步骤简单,便于应用;采用GC-ECD法同时测定了24种中药材中的10种农药残留,方法简便,易于操作。
农药残留量测定中,《中国药典》规定了有机氯类农药残留量的测定方法,包括六六六、滴滴涕和五氯硝基苯。规定甘草和黄芪的限量为总 BHC≤0.2×10-6,总 DDT≤0.2×10-6,PCNB≤0.1×10-6[5]。美、英和欧洲药典除规定了有机氯类农药残留量外,还规定了有机磷类农药的限量:毒死蜱0.2 ppm,敌敌畏1.0 ppm,六六六0.3 ppm,马拉硫磷1.0 ppm,对硫磷0.5 ppm,五氯硝基苯1.0 ppm,滴滴涕1.0 ppm[6~8],本文所测样品中的农药残留量均低于中、美、英及欧洲药典要求。
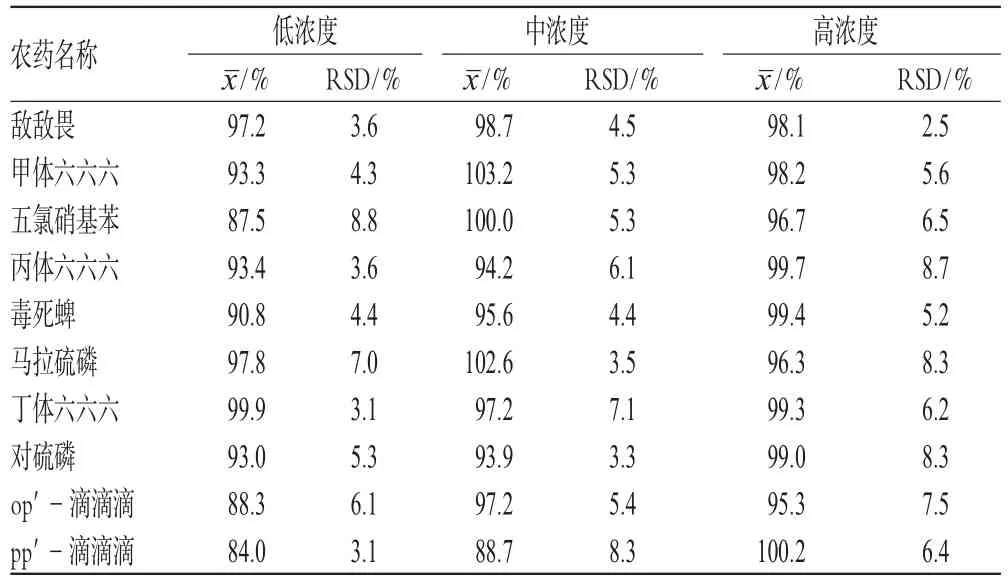
表3 回收率试验结果(n=3)Tab 3Results of recovery tests(n=3)
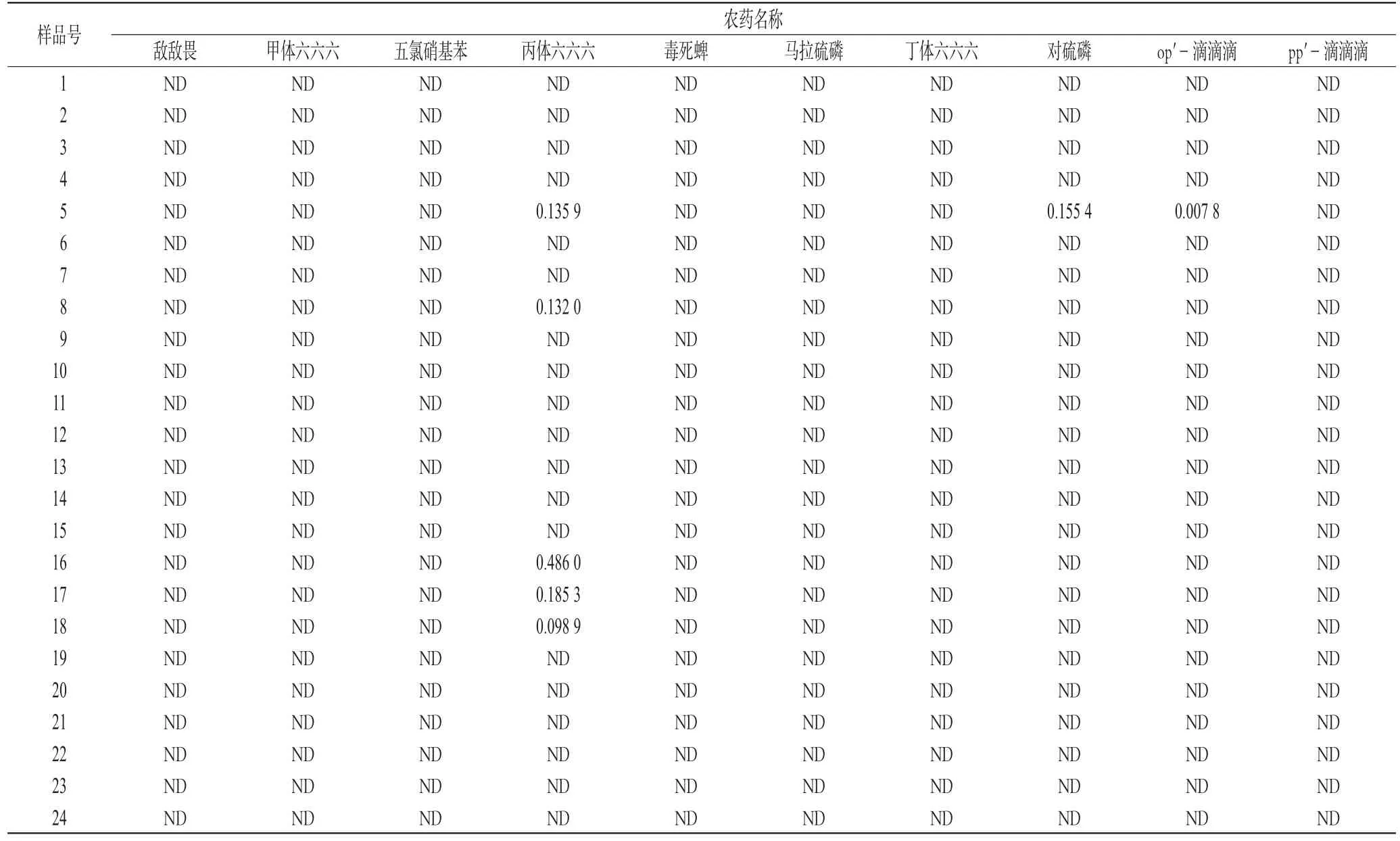
表4 样品中农药残留量(mg·kg-1)Tab 4 Pesticide residues in the samples( mg·kg-1)
[1] 万益群,李申杰,鄢爱平.白术中有机磷及氨基甲酸酯类农药残留量的测定[J].分析科学学报,2007,23(3):299.
[2] 向增旭,赵维佳,郭巧生.金银花中18种有机磷农药残留量分析方法的研究[J].中国中药杂志,2006,31(16):1 321.
[3] 马虹英.浅谈中药中有机氯农药残留问题[J].中国药房,2005,16(21):1 667.
[4] 周洪波,房志坚,杨立伟,等.固相萃取-GC-MS联用法测定何首乌中有机氯农药残留量[J].中国药房,2010,21(19):777.
[5] 国家药典委员会编.中华人民共和国药典(一部)[S].2010年版.北京:中国医药科技出版社,2010:附录57.
[6] The United States Pharmacopeial Convention.U.S.Pharmacopeia(24)/National Formulary(19)[S].Philadelphia PA:States Pharmacopeial Convention,2000:19.
[7] British Pharmacopoeia Commission.British Pharmacopoeia[S].London:States Pharmacopeial Commission,2000:Appendix,81.
[8] Council of Europe.European Pharmacopoeia[S].Strasbourg:States Pharmacopeial Convention,2011:242.
猜你喜欢
化工设计(2022年4期)2023-01-02
化工管理(2022年13期)2022-12-02
云南化工(2022年9期)2022-10-12
石油炼制与化工(2022年2期)2022-02-15
能源化工(2021年6期)2021-02-26
化工管理(2020年26期)2020-10-09
农药科学与管理(2019年12期)2019-05-20
中成药(2018年1期)2018-02-02
电子技术与软件工程(2016年24期)2017-02-23
中国药业(2014年24期)2014-05-26
