
口炎清颗粒指纹图谱研究*
2011-07-24 09:49关倩怡王德勤苏薇薇
中山大学学报(自然科学版)(中英文) 2011年1期
关倩怡,黄 琳,彭 维,王德勤,苏薇薇
(1.中山大学生命科学学院,广东 广州 510275;2.广州白云山和记黄埔中药有限公司,广东 广州 510515)
口炎清颗粒是由山银花、玄参、天冬、麦冬、甘草五味药组成的中药制剂(国家中药保护品种),具有清热养阴、解毒消肿的功效,用于治疗阴虚火旺所致的口腔溃疡[1],药效确切[2]。本研究建立了口炎清颗粒的指纹图谱,为其质量监控提供了依据,有利于确保该产品质量的稳定和均一。
1 仪器与试药
Dionex P680型高效液相色谱仪(四元梯度泵、自动进样器、ATH-585柱温箱、PDA-100检测器及Chromeleon工作站,美国戴安公司);BP211D电子分析天平(瑞士沙多利斯公司);T660/H超声波清洗器(美国埃玛公司)。
绿原酸(批号:110753-200413)、咖啡酸(批号:110885-200102)、木犀草苷(批号:111720-200602)、哈巴俄苷(批号:111730-200604)、肉桂酸(批号:110786-200503)、甘草酸铵(批号:110731-200614)、甘草苷(批号:110610-200604)等对照品,均购自中国药品生物制品检定所;口炎清颗粒及其半成品由广州白云山和记黄埔中药有限公司提供。
乙腈 (美国Burdick & Jackson) 为色谱纯;甲醇(广东光华化学厂有限公司)、甲酸(天津市富宇精细化工有限公司)均为分析纯;水为超纯水。
2 方法与结果
2.1 溶液的制备
2.1.1 对照品溶液的制备 分别精密称取绿原酸、咖啡酸、木犀草苷、哈巴俄苷、肉桂酸、甘草苷、甘草酸铵对照品约2 mg,置10 mL量瓶中,用甲醇溶解并定容至刻度,摇匀,制成每mL含绿原酸、咖啡酸、木犀草苷、哈巴俄苷、肉桂酸、甘草苷、甘草酸铵各0.2 mg的对照品溶液。
2.1.2 成品供试品溶液的制备 取口炎清颗粒10袋,研细,取约10 g,精密称定,置具塞锥形瓶中,精密加入甲醇50 mL,密塞,称定质量,超声处理(功率360 W,频率35 kHz)30 min,放冷,再称定质量,用甲醇补足减失的质量,摇匀,滤过,精密量取续滤液10 mL,减压回收溶剂至干,残渣加水约5 mL溶解,定量转移至固相萃取小柱(填料:C18,规格:6 mL,500 mg),加水15 mL分次淋洗,淋洗液弃去,再用甲醇15 mL分次洗脱,收集洗脱液,回收溶剂至干,残渣定量加入甲醇2 mL,使完全溶解,用0.45 μm微孔滤膜滤过,作为成品供试品溶液。
2.1.3 药材供试品溶液的制备 分别取药材粗粉,山银花1 g、玄参0.8 g、甘草0.5 g,精密称定,分别置于圆底烧瓶中,加入φ=50 %乙醇50 mL,回流提取2 h,滤过,滤液减压浓缩至干,残渣加水约5 mL溶解,定量转移至SPE固相萃取小柱(填料:C18,规格:6 mL,500 mg),加水15 mL分次淋洗,淋洗液弃去,再用甲醇15 mL分次洗脱,收集洗脱液,回收溶剂至干,残渣用少量甲醇溶解,定容至10 mL,用0.45 μm的微孔滤膜滤过,作为药材供试品溶液。
2.1.4 阴性供试品溶液的制备 分别取缺山银花、缺玄参、缺甘草的阴性样品适量,按“2.1.2 成品供试品溶液的制备”的方法操作。
2.2 色谱条件
Dikma PLATISIL ODS(250 mm×4.6 mm,5 μm)色谱柱;以乙腈为流动相A,以φ=0.1%甲酸溶液为流动相B,梯度洗脱:0~15 min,B(98→90);15~120 min,B(90→59);120~125 min,B(59→59);流速:0.8 mL/min;检测波长:254 nm;柱温:25 ℃。理论塔板数以绿原酸计算不低于4 000。
3 方法学考察
3.1 精密度试验
按“成品供试品溶液的制备”方法制备口炎清颗粒供试品溶液,连续进样6次,记录HPLC色谱图,以保留时间28 min峰面积较大、较稳定的2号色谱峰(绿原酸)作为参照峰,计算各特征峰相对保留时间和相对峰面积的RSD。结果相对保留时间的RSD为0.05 %~0.20%,相对峰面积的RSD为0.48 %~1.78 %;采用《中药色谱指纹图谱相似度评价系统2004A版》进行评价,相似度大于0.999,表明精密度好。
3.2 稳定性考察
按“供试品溶液的制备”方法制备口炎清颗粒供试品溶液,分别在0、3、6、9、12、24、48 h进样分析,记录HPLC色谱图,以绿原酸色谱峰作为参照峰,计算各特征峰相对保留时间和相对峰面积的RSD。结果相对保留时间的RSD为0.05 %~0.44 %,相对峰面积的RSD为0.38 %~1.93 %;采用《中药色谱指纹图谱相似度评价系统2004A版》进行评价,相似度大于0.999,表明供试品溶液在放置48 h内稳定性良好。
3.3 重复性试验
取同一批口炎清颗粒,按“成品供试品溶液的制备”方法平行操作,制备6份口炎清颗粒供试品溶液,分别进样分析,记录HPLC色谱图,以绿原酸色谱峰作为参照峰,计算各特征峰相对保留时间和相对峰面积的RSD。结果相对保留时间的RSD为0.09 %~0.34 %,相对峰面积的RSD为0.19 %~2.04 %;采用《中药色谱指纹图谱相似度评价系统2004A版》进行评价,相似度大于0.980,表明重复性好。
4 口炎清颗粒指纹图谱的构建及相关技术参数
4.1 指纹图谱的构建
取10个批号的口炎清颗粒供试品溶液,按“2.2”项下色谱条件进行HPLC分析,记录色谱图,见图1。10批口炎清颗粒共检测到 23个共有特征峰,通过《中药色谱指纹图谱相似度评价系统2004 A版》对10批口炎清颗粒高效液相指纹图谱进行评价,获得共有模式(参照指纹图谱)见图2。
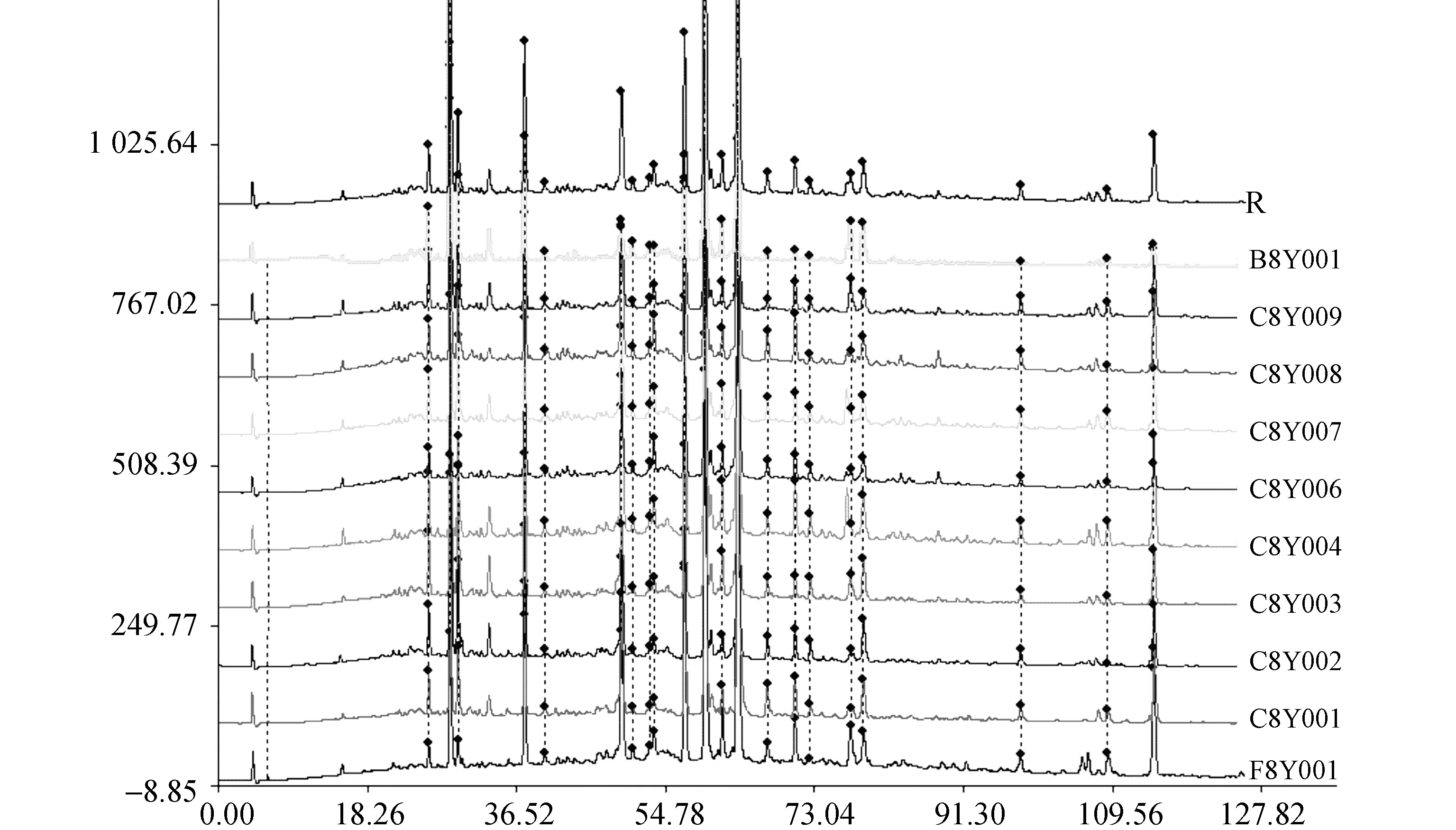
图1 10批口炎清颗粒HPLC指纹图谱
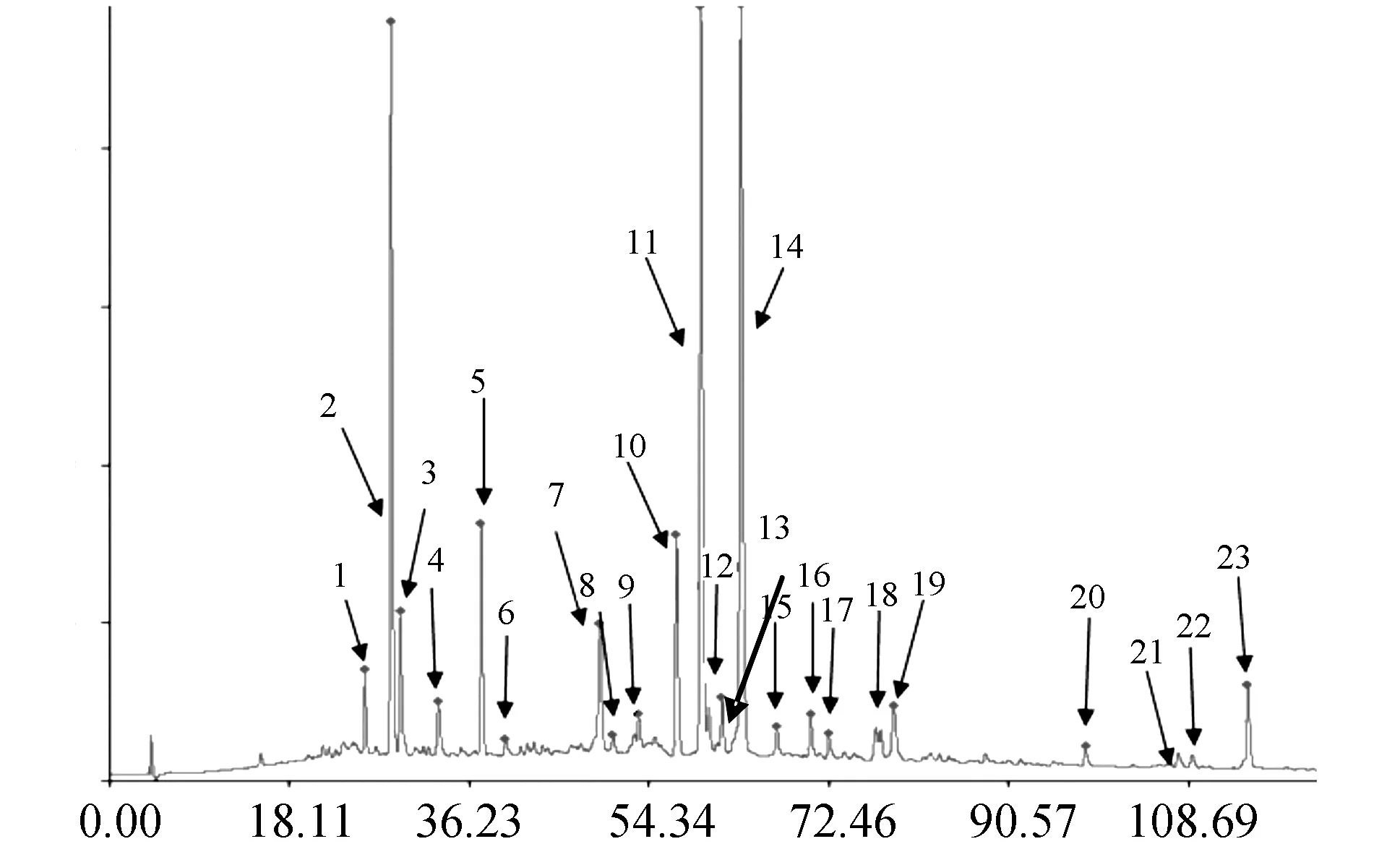
图2 口炎清颗粒HPLC参照指纹图谱
4.2 10批口炎清颗粒指纹图谱比较
10批口炎清颗粒相似度系数均大于0.90,证明口炎清颗粒的生产工艺稳定,产品的均一性较好。
4.3 共有峰的标定及归属
分别精密吸取药材及阴性供试品溶液,注入液相色谱仪,采集色谱图。通过保留时间及PDA光谱对比,确定口炎清颗粒23个共有特征峰中,归属于山银花的色谱峰应有12个,归属于玄参的色谱峰有4个,归属于甘草的色谱峰应有8个,其中18号峰为玄参与甘草共有,见图3~5。取对照品溶液 ,按“2.2”项下色谱条件,进样分析,根据保留时间定性,采用PDA光谱对比,确定2、4、7、9、18、19、23号峰分别为绿原酸、咖啡酸、甘草酸铵、木犀草苷、哈巴俄苷、肉桂酸、甘草苷,结果见图6。

图3 口炎清颗粒指纹图谱、山银花药材色谱图及缺山银花阴性色谱图
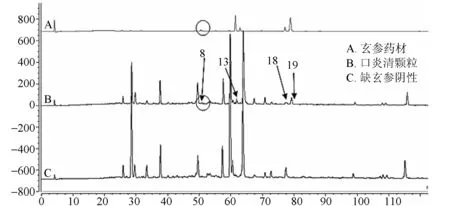
图4 口炎清颗粒指纹图谱、玄参药材色谱图及缺玄参阴性色谱图

图5 口炎清颗粒指纹图谱、甘草药材色谱图及缺甘草阴性色谱图
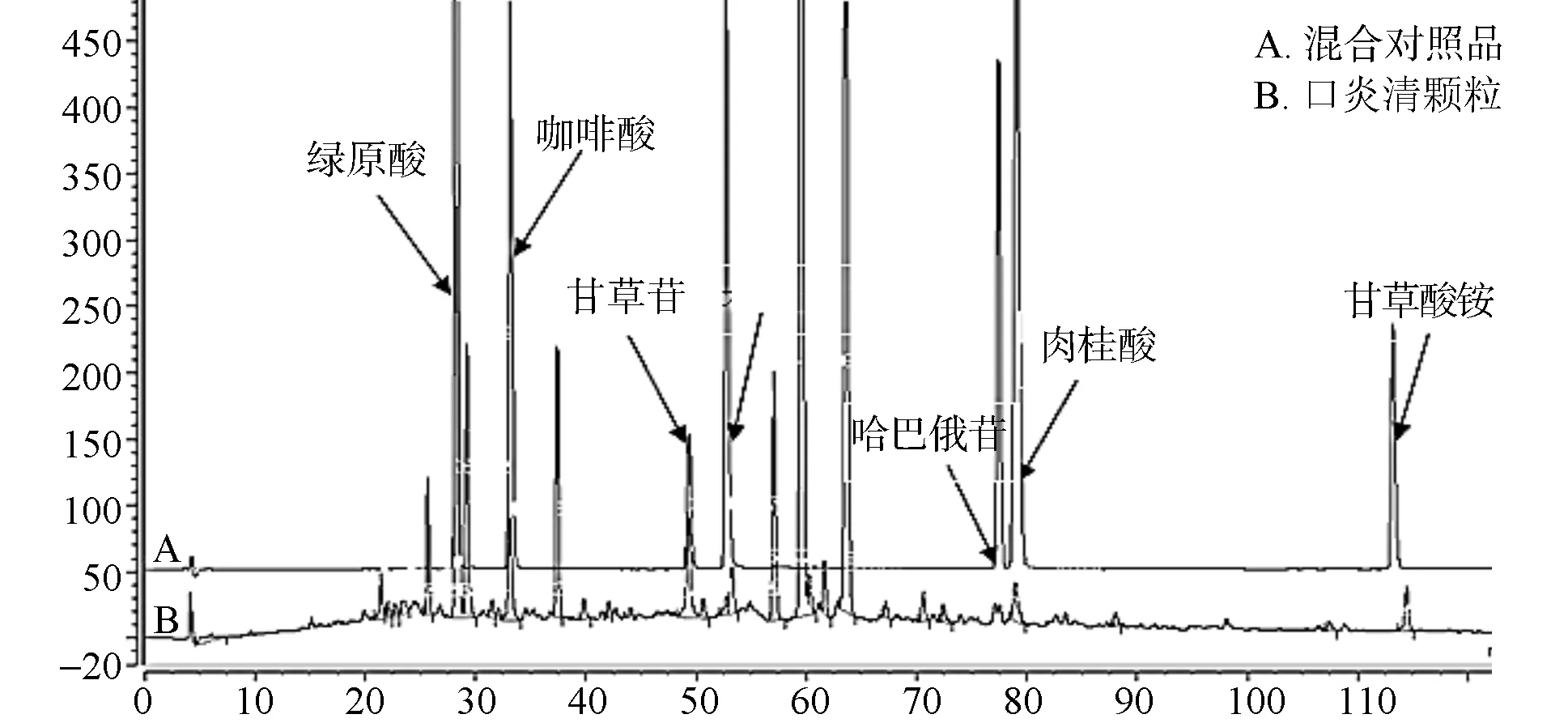
图6 口炎清颗粒指纹图谱及混合对照品色谱图
5 讨 论
1) 口炎清颗粒含有机酸、环烯醚萜、多羟基黄酮等成分[3-5],这些成分易溶于水,在色谱柱上不被保留,导致出峰时间太快。因此,选择流动相时,在水相中加入φ=0.1%甲酸,可有效控制出峰时间,使指纹图谱各色谱峰分离效果良好。
2) 采用二极管阵列检测器 (PDA),考察了200~400 nm范围波长的图谱,分别在波长254、278、290、355、208 nm测试样品[6-8],254 nm能涵盖绝大多数的峰,基线平稳,分离度良好。虽然绿原酸、咖啡酸、甘草苷在此紫外吸收较低,但其在口炎清颗粒中的含量大,仍能被检出,最终综合考虑选用254 nm作为检测波长。
3) 通过对照品加入法及PDA光谱定性,分别确证了绿原酸、咖啡酸、木犀草苷、哈巴俄苷、肉桂酸、甘草酸铵、甘草苷7个已知成分的色谱峰。本研究构建的指纹图谱,操作简便,专属性强、重复性好,为该产品的质量监控提供了有效手段,有利于确保产品质量的稳定、均一。
参考文献:
[1] 中华人民共和国药典委员会.中国药典:一部[M].北京:化学工业出版社,2005:334.
[2] 李忠思,张小娜,梁永,等.口炎清药效学研究.中药新药与临床药理, 1999, 10(4):216-217.
[3] 柴兴云, 李萍, 唐力英.山银花化学成分研究[J].中国中药杂志,2004, 29(9):865-866.
[4] 赵国玲,刘佳佳,林丹,等.金银花化学成分及药理研究进展[J].中成药,2002,24(12):973-976.
[5] 师怡,许晖,阙慧卿,等.玄参化学成分的药理作用和分析方法[J].海峡药学,2006,18(4):58-60.
[6] 张宇平,黄可龙.高效液相色谱法同时测定金银花中5种有机酸[J]. 分析试验室,2007,26(7):68-69.
[7] 刘承伟,毕志明,祝艳斐,等.玄参中4种主要活性成分的HPLC定量分析[J].中国药学杂志,2007,42(21):1614-1616.
[8] 吴昭晖,罗佳波,游文玮.甘草药材HPLC 指纹图谱研究[J].中草药,2005,36(12):1868-1872.
猜你喜欢
今日农业(2021年21期)2021-11-26
中老年保健(2021年9期)2021-08-24
现代畜牧科技(2021年4期)2021-07-21
天然产物研究与开发(2019年1期)2019-03-01
中成药(2018年11期)2018-11-24
天然产物研究与开发(2018年7期)2018-08-21
中成药(2017年10期)2017-11-16
中国卫生标准管理(2015年8期)2015-01-26
中医研究(2014年2期)2014-03-11
中国中医药现代远程教育(2014年20期)2014-03-01
