
Aβ诱导内质网应激性凋亡通路的启动及二苯乙烯苷的影响
2011-06-22 01:04罗红波杨金升石向群李芸杨期东张志强尹榕
东南大学学报(医学版) 2011年6期
罗红波,杨金升,石向群,李芸,杨期东,张志强,尹榕
(1.兰州军区总医院神经内科,甘肃兰州 730050;2.中南大学湘雅医院 神经内科,湖南 长沙 410003)
阿尔茨海默病(Alzheimer's disease,AD)是一种神经系统变性疾病,其主要的病理学特征之一是脑组织中出现大量老年斑[1]。β-淀粉样蛋白(β-amyloid peptide,Aβ)是老年斑的主要成分,因此Aβ的神经毒性是AD形成和发展的关键因素,其诱发的神经元变性和凋亡与AD患者认知功能、行为学障碍密切相关。近来又发现内质网功能改变如内质网应激可导致蛋白质分子构象功能障碍,使蛋白质无法与输送机制耦联,从而引发疾病[2];同时,内质网应激介导Caspase12活化,由于Caspase12是内质网应激反应中特异性关键蛋白酶[3],内质网应激介导Caspase12活化是否参与AD发病中细胞凋亡的发生国内外少见报道。本实验通过建立AD动物模型,研究在Aβ1-42诱导大鼠学习记忆障碍发生发展中海马神经元Caspase12表达水平的变化及其在介导神经元凋亡中的作用。
1 材料和方法
1.1 材料
1.1.1 实验动物
选用健康4月龄Wistar大鼠80只(中南大学实验动物中心提供),雌雄各半,体重250~350 g。
1.1.2 主要试剂和药物
Caspase12一抗、Aβ 1-42购于Santa Cruz公司;引物自行设计,由上海华大基因有限公司合成;Trizol、Taq酶、逆转录酶购自MBI公司。TSG为从何首乌中提取分离的干粉,含量为68%(湖南中医药研究所提供),实验时用水溶解为100 mg·kg-1的浓缩液。
1.2 方法
1.2.1 大鼠分组、模型建立及干预
将选择好的动物应用随机数字表进行分组,分为对照组、假手术组、模型组、TSG组各20只。模型组:水合氯醛按2.5 ml·kg-1腹腔麻醉,立体定向仪下固定头部,参考大鼠图谱,确定前囟后3.0 mm、中线旁2.5 mm为海马所在部位,钻破颅骨,微量注射器进针约2.9 mm,左右两侧各缓慢均匀注入 Aβ 1-42各5 μg,术后缝合皮肤,正常饲养大鼠,灌胃液为生理盐水。对照组:正常饲养大鼠,灌胃液为生理盐水;假手术组:注射液为等量生理盐水,灌胃液为生理盐水;TSG组:注射液为Aβ 1-42,灌胃液为TSG悬液。各组于制模前及注射后3、21 d 3个时间点宰杀取材,每组6只取海马组织,一侧供提取内质网蛋白,另一侧供提取RNA。以上各组动物于造模前3 d开始给药,每天灌胃1次,动物灌胃量按5 ml·kg-1计算。
1.2.2 行为学检测
1.2.2.1 Y-电迷宫学习记忆实验 电击次数表示其学习记忆的获得能力,记录每只大鼠学会逃避电刺激次数,以30次为最大值计数。行为学测试分别于制模前3 d、制模后3 d及21 d进行。
1.2.2.2 Morris水迷宫定位航行实验 每天8:00到11:00之间对每只大鼠进行训练,不选平台所在象限作为入水点,按顺时针方向把大鼠面向池壁放入水中,检测大鼠隐匿平台潜伏期。如果在规定的最长时间120 s内未到达平台,则由实验者将其引至平台,逃避潜伏期记为120 s,大鼠在平台休息20 s后进行下一次测试。观察并记录动物找到并爬上平台的潜伏期、游泳路程及游泳轨迹。3 d训练结束后将大鼠按组别进行造模,分别于制模前3 d、制模后3 d及21 d进行实验。
1.2.2.3 Morris水迷宫空间探索试验 各组大鼠最后一次定位航行实验后撤除平台,将大鼠从最后1次入水点面向池壁放入水中,使大鼠在水迷宫中连续游泳,记录大鼠120 s内穿越原平台平面的次数。
1.2.3 海马部位神经元凋亡率测定
采用罗氏公司原位凋亡检测试剂盒检测,方法为TUNEL法。用Olympus显微镜观察各组细胞凋亡变化趋势,棕黄色细胞为阳性凋亡细胞,无显色为阴性,随机计数10个视野中阳性、阴性细胞。光镜下计算凋亡指数(apoptosis index,AI)。计算方法:每只动物随机两张切片,每张切片分别在海马CA1区观察大小为1 mm的2个区域,分别计算各区凋亡细胞数和总细胞数,AI=凋亡细胞数/总细胞数×100/%。
1.2.4 逆转录聚合酶链反应(RT-PCR)测 mRNA表达
依据Medline基因文库自行设计引物,目的基因Caspase12(363 bp),正义链:5'-CAATTCTAACTGTC CGAGTCTGAG-3',反义链:5'-CTATCGGTGACGAC TATGTCTACT-3';内参照β-actin(100 bp),正义链:5'-CGTTGACATCCGTAAAGACCTCTA-3',反义链:5'-TAAAACGCAGCTCAGTAACAGTCCG-3'。产物经琼脂糖凝胶电泳,于紫外投射灯下观察结果并拍照。
1.2.5 免疫印迹法(Western blotting)测蛋白表达
密度梯度离心法提取内质网蛋白,将海马组织转移到匀浆器中,加裂解液匀浆,离心(2 400 r·min-1,10 min),取上清,继续离心(12 000 r·min-1,15 min),取上清,再离心(15 000 r·min-1,30 min),加入裂解液和蛋白酶抑制剂,用BCA蛋白测定试剂盒测定蛋白质浓度。样品与等体积的上样缓冲液混匀,煮沸变性,用10%SDS-PAGE分离,转移蛋白至硝酸纤维素膜,5%脱脂奶粉封闭,与一抗孵育,加入辣根过氧化物酶标记的二抗孵育,ECL试剂盒进行化学发光检测,JS-300凝胶图像仪扫描分析处理。
1.3 统计学处理
2 结 果
2.1 Y-电迷宫检测
制模前各组间大鼠学会躲避所需的电刺激次数比较,F=0.860,P=0.429,说明各组间大鼠学习记忆能力差异无统计学意义,有可比性。制模后各组间大鼠学会躲避所需的电刺激次数比较,F=13.866,P=0.000,差异有统计学意义,说明制模成功。经TSG干预,TSG组制模后21 d学会躲避所需的电刺激次数减少,与模型组比较,t=13.126 3,P=0.000 0,差异有统计学意义。见表1。
表1 Y-电迷宫测试比较大鼠学习记忆能力±s)Tab 1 Learning capacity of rats by Y-maze±s)
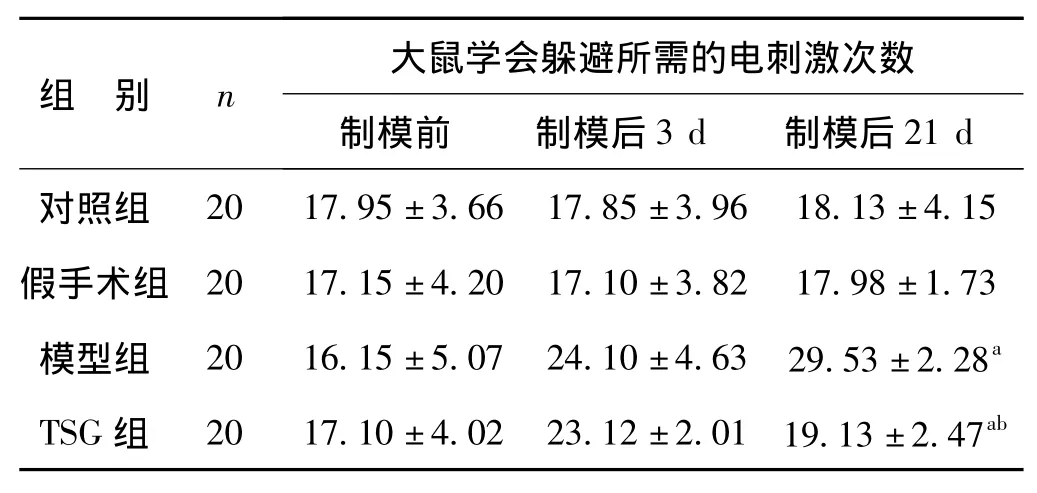
表1 Y-电迷宫测试比较大鼠学习记忆能力±s)Tab 1 Learning capacity of rats by Y-maze±s)
与对照组比较,a P<0.05;与模型组比较,b P<0.05
组 别 n 大鼠学会躲避所需的电刺激次数制模前 制模后3 d 制模后21 d对照组20 17.95±3.66 17.85±3.96 18.13±4.15假手术组 20 17.15±4.20 17.10±3.82 17.98±1.73模型组 20 16.15±5.07 24.10±4.63 29.53±2.28a TSG组 20 17.10±4.02 23.12±2.01 19.13±2.47ab
2.2 Morris水迷宫检测
与对照组相比,模型组和TSG组大鼠造模后3 d及21 d的Morris水迷宫测试游泳潜伏期及路程均增加(P<0.01),穿越平台次数下降明显(P>0.05),差别随时间的延长而加大,以21 d差别最明显(P<0.01);与模型组比较,TSG组3 d及21 d的潜伏期缩短(P<0.05),游泳路程缩短(P<0.05),穿越平台次数增加(P<0.05),差异具有统计学意义。假手术组与对照组比较,差异无统计学意义(P>0.05),排除手术因素干扰。见表2~4。
表2 水迷宫潜伏期测试比较大鼠空间分辨能力(±s)Tab 2 Space capacity of rats by Morris water maze'latency±s) s

表2 水迷宫潜伏期测试比较大鼠空间分辨能力(±s)Tab 2 Space capacity of rats by Morris water maze'latency±s) s
与对照组比较,a P<0.01;与模型组比较,b P<0.05
组 别 n 造模后不同时间游泳潜伏期0 d 3 d 21 d对照组20 17.81±1.09 18.05±1.96 17.03±1.23假手术组 20 18.10±1.16 17.93±1.32 17.87±1.07模型组 20 17.05±1.03 26.23±3.45a32.15±4.51a TSG组 20 18.03±1.12 22.06±2.52ab25.26±3.76ab
表3 水迷宫路程测试比较大鼠空间记忆能力±s)Tab 3 Space capacity of rats by Morris water maze'swimming distanc(±s) 米
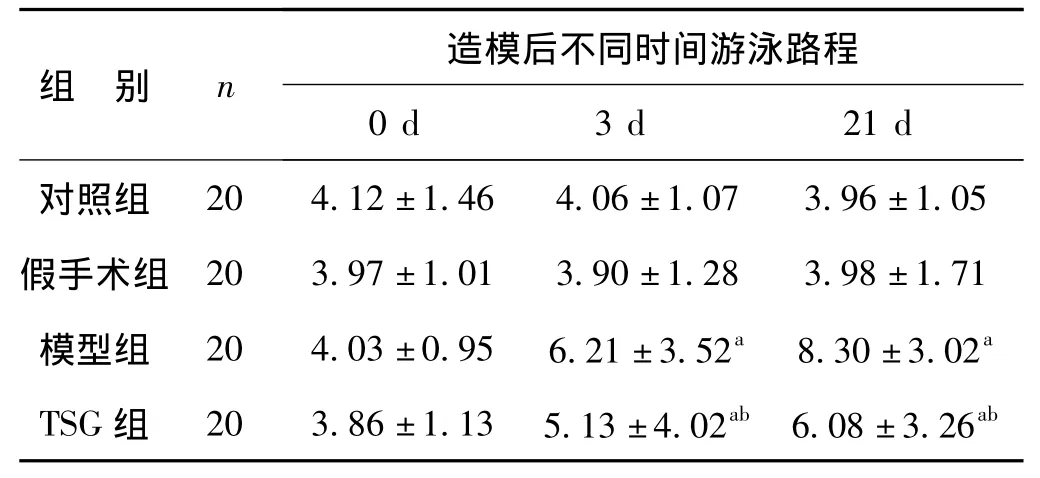
表3 水迷宫路程测试比较大鼠空间记忆能力±s)Tab 3 Space capacity of rats by Morris water maze'swimming distanc(±s) 米
与对照组比较,a P<0.01;与模型组比较,b P<0.05
组 别 n 造模后不同时间游泳路程0 d 3 d 21 d对照组20 4.12±1.46 4.06±1.07 3.96±1.05假手术组 20 3.97±1.01 3.90±1.28 3.98±1.71模型组 20 4.03±0.95 6.21±3.52a 8.30±3.02a TSG组 20 3.86±1.13 5.13±4.02ab 6.08±3.26ab
表4 水迷宫穿越平台次数测试比较大鼠工作记忆能力±s)Tab 4 Work capacity of rats by Morris water maze'crossing the exact former platform±s)

表4 水迷宫穿越平台次数测试比较大鼠工作记忆能力±s)Tab 4 Work capacity of rats by Morris water maze'crossing the exact former platform±s)
与对照组比较,a P<0.05;与模型组比较,b P<0.05
组 别 n 造模后不同时间穿越平台次数0 d 3 d 21 d对照组20 6.05±2.13 6.15±2.35 18.13±4.15假手术组 20 5.95±3.17 6.01±1.91 17.98±1.73模型组 20 6.12±1.09 4.34±1.21a 3.09±1.18a TSG组 20 6.06±2.06 5.14±1.03ab 4.82±1.09ab
2.3 大鼠海马CA1区的凋亡率测定
光镜下观察,Tunel染色阳性凋亡细胞胞核呈深棕色,细胞体积变小,成三角形或扇形,尼氏小体消失,核固缩深染,阴性的细胞核则不显示棕色,细胞形态正常,较易区别。对照组大鼠海马CA1区只有零星的凋亡细胞,而模型组大鼠CA1区可见大量的凋亡细胞,TSG组凋亡细胞较模型组明显减少(图1)。对照组、模型组、TSG组细胞凋亡率比较差异有统计学意义(P<0.05或P<0.01)(表5)。假手术组与对照组比较差异无统计学意义(P>0.05),排除手术因素干扰。

图1 光镜下大鼠海马CA1区Tunel法阳性凋亡细胞Fig 1 Positive cells of CA1 region in rats hippocampus by Tunnel under light microscope
表5 大鼠海马CA1区凋亡率比较±s)Tab 5 Apoptosis rates of CA1 region in rats hippocampus±s)

表5 大鼠海马CA1区凋亡率比较±s)Tab 5 Apoptosis rates of CA1 region in rats hippocampus±s)
与对照组比较,a P<0.01,与模型组比较,b P<0.05
组 别 n CA1区凋亡率/%对照组20 2.46±1.12假手术组 20 2.39±1.08模型组 20 18.42±2.18a TSG组 20 12.97±1.24b
2.4 内质网凋亡因子 Caspase12的 mRNA及蛋白表达
模型组的Caspase12 mRNA与其蛋白的表达趋势一致,Caspase12在3 d表达增加,然后逐渐下降,21 d但仍有少量表达;0、3、21 d的Caspase12的蛋白表达分别为对照组的99.3%、133.3%和118.6%,各时间点差异有统计学意义(F=54.00,P=0.00)。TSG组Caspase12在3 d的表达强度明显较弱,与模型组比较差异有统计学意义(t=2.717 5,P=0.021 7);假手术组与对照组比较差异无统计学意义(t=0.679 4,P=0.512 3),排除手术因素干扰。见表6,图2、3。
3 讨 论
AD作为老年常见疾病之一,以早期出现记忆力下降为主要表现,其中老年斑是AD的主要病理特征之一,而纤维化的Aβ是构成老年斑的主要成分[4]。目前发现Aβ形成过程中的神经毒性可引起神经元的内质网等细胞亚结构发生变性,细胞的凋亡最终导致脑萎缩等脑组织形态学改变[5-6]。因此,我们在大鼠脑内双侧海马区注入Aβ 1-42,观察Aβ 1-42神经毒性对大鼠行为学及细胞内质网功能的影响及TSG的干预作用。
我们对何首乌的相关研究表明,何首乌能改善AD模型大鼠学习记忆能力,对胆碱能神经投射纤维有保护作用,可提高乙酰胆碱酯酶活性,可通过调节凋亡相关基因Bax/Bcl-2通路抑制细胞凋亡[7-11]。因此,我们对何首乌的主要有效成分TSG进行研究,进一步揭示其治疗AD的主要作用机制及药物靶点。Y电迷宫实验主要考察大鼠学习记忆保持能力,水迷宫实验中,定位航行实验主要考察大鼠的空间分辨学习能力,空间探索实验主要测量大鼠的工作记忆能力。通过本实验可以看出,在Aβ毒性作用下,模型大鼠的学习记忆、空间定向、工作记忆能力明显下降,表现为Y电迷宫躲避所需的电刺激次数增加,Morris水迷宫测试中潜伏期延长,游泳路程增加及穿越平台次数减少;从时间上来看,这种损害随着Aβ暴露时间的延长而加重,说明Aβ对大鼠的学习记忆、空间记忆及工作记忆的损害呈逐渐增强的累加效应。经TSG干预后,大鼠躲避所需的电刺激次数减少,潜伏期缩短,游泳路程缩短,穿越平台次数增加,说明TSG可改善模型大鼠学习记忆、空间定向、工作学习能力,具有脑保护作用。
表6 及蛋白相对表达量(Caspase 12/β-actin) (±s)Tab 6 The mRNA and protein expression of Caspase12±s)

表6 及蛋白相对表达量(Caspase 12/β-actin) (±s)Tab 6 The mRNA and protein expression of Caspase12±s)
与对照组比较,a P <0.05,b P >0.05,c P<0.01;与模型组比较,d P <0.05
组 别 n mRNA相对表达/%蛋白相对表达量/%.24±0.02 0.20±0.02假手术组 20 0.20±0.02 0.21±0.03b 0.19±0.01 0.24±0.02 0.25±0.03b 0.21±0.01模型组 20 0.19±0.01 0.27±0.02c 0.24±0.02 0.23±0.02 0.32±0.02c 0.26±0.02 TSG组 20 0.20±0.02 0.22±0.03d 0.21±0.02 0.23±0.02 0.26±0.03d 0 d 3 d 21 d对照组 20 0.21±0.01 0.19±0.02 0.20±0.02 0.23±0.01 0 0 d 3 d 21 d 0.23±0.02
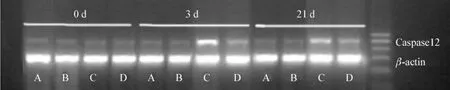
图2 Caspase12的mRNA表达Fig 2 The mRNA expression of Caspase12

图3 Caspase12/β-actin的蛋白表达Fig 3 The protein expression of Caspase12 and β-actin
内质网是蛋白质合成、加工和转运的主要场所,在真核细胞的生长发育中起着关键作用。内质网腔内有大量的“分子伴侣”,辅助和监控蛋白质的正确折叠并装配成天然构象。在某些刺激因素下,内质网的功能受到影响,造成大量未折叠或错误折叠的蛋白质堆积,由此引起一系列“分子伴侣”表达上调,以帮助蛋白进一步正确折叠、装配,使受损细胞在应激情况下存活,这个应答过程称为内质网应激[11]。内质网应激反应性细胞凋亡是不同于受体介导或经线粒体介导的一种新的细胞凋亡途径。Caspase12作为凋亡起始因子发挥了关键作用[13]。Caspase12酶原位于内质网膜侧,活化后可独立地诱导内质网特异性的细胞凋亡[14]。
既往报道认为在AD发病机制中,Aβ可破坏细胞膜,形成大量自由基,启动死亡受体及线粒体途径导致神经元的凋亡[15-17]。本研究发现模型鼠海马组织有内质网特异的Caspase12基因和蛋白高表达,说明Aβ还可以引起内质网应激活化Caspase12,启动内质网应激介导的神经元凋亡信号通路,导致神经元死亡,发生学习记忆障碍。当Aβ给药3 d后,Caspase12的表达升高,推测内质网为保护受损细胞而首先选择程序性细胞死亡,启动了特异性的Caspase12凋亡通路;Caspase12在21 d表达下降可能与以下因素有关:内质网应激后ATF6信号传导通路激活,GRP78表达增多,减少错误折叠的蛋白使内质网应激的程度下降[18],同时,GRP78可与 Caspase7形成复合物,阻止Caspase12激活并阻止其从内质网释放[19];随着时间的延长,内质网应激程度逐渐下降,至21 d后消失,相应的凋亡途径也被抑制,但已经造成的组织损害仍持续存在(如老年斑),可能对诱发神经元凋亡起一定的作用[20]。假手术组各时间段与对照组比较无显著性差异,表明手术作为一过性短暂的干扰因素,对本实验研究结果不产生影响。
因此,内质网应激及相关的凋亡通路可能参与Aβ神经毒性对脑的神经元损伤。在AD的发病中,当Aβ被细胞分泌至细胞外形成纤维化的过程中,所表现的神经毒性开始起作用,细胞外的Aβ可破坏细胞膜诱发氧化应激,氧化应激又会破坏内质网功能引起内质网应激,应激过度导致通过特异性的内质网Caspase12凋亡途径发生死亡;同时内质网的应激也使细胞内产生的Aβ在内质网中错误折叠的几率增加,既导致了Aβ的异常剪切和积聚,又加剧了氧化应激和内质网功能障碍。在上述诸因素的相互作用下,形成一种恶性循环,使损害效应不断扩大,最终导致神经元凋亡,出现AD的病理学改变及进行性加重的行为学障碍。
[1]QUERFURTH H W,LAFERLA F M.Alzheimer's disease[J].N Engl J Med,2010,362(4):329-344.
[2]AUSAIN R C.The unfolded protein response in health and disease[J].Antioxid Redox Signal,2009,11(9):2279-2287.
[3]RASHEVA V I,DOMINGOS P M.Cellular responses to endoplasmic reticulum stress and apoptosis[J].Apoptosis,2009,14(8):996-1007.
[4]TOMIYAMA T.Involvement of beta-amyloid in the etiology of Alzheimer's disease[J].Brain Nerve,2010,62(7):691-699.
[5]SALMINEN A,KAUPPINEN A,SUURONEN T,et al.ER stress in Alzheimer's disease:a novel neuronal trigger for inflammation and Alzheimer's pathology[J].J Neuroinflammation,2009,26(6):41-46.
[6]秦锦标.基于体素的形态测量学在阿尔茨海默病中的应用[J].东南大学学报:医学版,2010,29(4):21-23.
[7]侯德仁,杨期东,周琳,等.何首乌对Alzhemer病模型大鼠学习记忆的影响及其机制的研究[J].中国医师杂志,2004,6(3):347-349.
[8]李吴,杜小平,杨期东,等.何首乌对海人藻酸致大鼠脑Ach能神经元及纤维损伤的保护作用[J].卒中与神经疾病,2002,9(5):299-302.
[9]周琳,杨期东,马志健,等.何首乌对Alzheimer病大鼠学习记忆及乙酞胆碱酷酶的作用[J].中风与神经疾病杂志,2004,21(5):394-396.
[10]周琳,杨期东,袁梦石,等.何首乌制剂在致海马神经元凋亡中的Bax表达[J].中风与神经疾病杂志,2006,23(3):143-145.
[11]罗红波,杨期东,鲍娟,等.何首乌提取物二苯乙烯苷对Alzheimer病模型大鼠学习记忆及海马超微结构的影响[J].中国行为医学科学,2008,17(5):402-403.
[12]HOROWITZ J C,LIMPER A H.Stress in the ER(endoplasmic reticulum):a matter of life and death for epithelial cells[J].Am J Respir Crit Care Med,2008,178(8):838-846.
[13]NAKAGAWA T,YUAN J.Cross-talk between two cysteine protease families activation of caspase-12 by calpain in apoptosis[J].Journal of Cell Biology,2000,150(4):887-894.
[14]JUNICHI H,TAIICHI K,MANABU T.Apoptosis induced by endoplasmic reticulum stress depends on activation of caspase-3 via caspase-12[J].Neuroscience Letters,2004,357(2):127-130.
[15]SHIBATA N,KOBAYASHI M.The role for oxidative stress in neurodegenerative diseases[J].Brain Nerve,2008,60(2):157-170.
[16]LEUNER K,PANTELl J,FREY C,et al.Enhanced apoptosis,oxidative stress and mitochondrial dysfunction in lymphocytes as potential biomarkers for Alzheimer's disease[J].J Neural Transm Suppl,2007,72(7):207-215.
[17]HAUPTMANN S,KEIL U,SCHERPING I,et al.Mitochondrial dysfunction in sporadic and genetic Alzheimer's disease[J].Exp Gerontol,2006,41(7):668-673.
[18]DOROUDGAR S,THUERAUF D J,MARCINKO M C,et al.Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response[J].J Biol Chem,2009,284(43):29735-29745.
[19] HAYASHI T,SAITO A,OKUNO S,et al.Induction of GRP78 by ischemic preconditioning reduces endoplasmic reticulum stress and prevents delayed neuronalcell death[J].Cereb Blood Flow Metab,2003,23(8):949-961.
[20]KIM I,XU W,REED J C.Cell death and endoplasmic reticulum stress:disease relevance and therapeutic opportunities[J].Nat Rev Drug Discov,2008,7(12):1013-1030.
猜你喜欢
信息记录材料(2022年11期)2023-01-07
解放军医学杂志(2021年12期)2022-01-18
现代临床医学(2021年1期)2021-01-26
铸造设备与工艺(2018年2期)2018-05-22
文理导航·科普童话(2016年7期)2017-02-04
安徽医科大学学报(2016年12期)2017-01-15
文理导航·科普童话(2016年4期)2016-05-31
儿童故事画报·智力大王(2015年12期)2016-01-23
锻压装备与制造技术(2015年3期)2015-08-15
儿童故事画报·智力大王(2015年2期)2015-05-20
