
大鼠脑出血灶周水肿变化与缺血改变的研究
2011-06-01 09:56牛小媛
中国医药导报 2011年9期
胡 明,牛小媛
山西医科大学第一附属医院神经内科,山西太原 030001
脑出血是神经科常见病,在常规治疗后,大约有1/3的脑出血患者在发病后一段时间内仍出现进行性神经功能恶化,提示除血肿引起的急性神经组织损害外,还存在有血肿周边的继发性损害。这部分患者在影像学上表现为持续存在的环形低密度影,对于此区域的认识集中在水肿带和缺血梗死带两方面。脑水肿主要与细胞内外异常液体积聚有关,主要分成血管源性脑水肿和细胞毒性脑水肿,由于病变的程度以及病情发展的不同,这两类脑水肿常常同时存在[1-2];Forbes等[3]对出血灶区域的DWI及MRIT2加权像研究发现:血肿周围除水肿外还是存在缺血带。可见对于此区域水肿与缺血共存可以达到共识,但水肿、缺血随时间的变化关系,至今还未有很明确的答案。本实验通过比较不同时间点大鼠ICH模型血肿灶周的水肿与缺血的变化过程,探讨大鼠脑出血水肿高峰期后灶周水肿及缺血的变化。
1 材料与方法
1.1 材料
1.1.1 实验动物 SD雄性大鼠,体重200 g左右,由山西医科大学动物实验中心提供。
1.1.2 试剂 2%TTC溶液、TUNEL试剂盒均购于山西省太原市boster试剂公司。
1.1.3 仪器 立体定位仪、必要手术器械;方正V70+扫描仪、计算机设备及软件。
1.2 动物模型制作
大鼠术前8 h禁食,不禁水。参照 Deinsberger[4]、周中和等[5]的报道方法,采用二次注血/退针法:缓慢注射自体血100μl至基底节区制成大鼠ICH模型;对照组相同部位打孔进针,不注血。实验阳性大鼠选择参照Bederson等[6]的方法:神经功能缺陷评分≥2分,及大鼠脑切片中有明显的圆形、椭圆形或不规则的血肿存在为模型成功标准,血液针道反流、进入脑室或死亡者均剔除。
1.3 试验分组
SD大鼠45只,30只造模。按起病后不同时间各分为3、5、7、14、21 d 5 个组,每组各 9 只(3 只对照+6 只模型)。
1.4 脑组织HE染色及TTC染色
各组大鼠于ICH损伤后在相应的时间点用过量的水合氯醛麻醉,直接断头取脑,完整取出脑组织,用滤纸吸干脑组织表面的液体和血迹,-20℃冰箱冷冻20~30 min,以进针点所在且垂直于大脑纵裂的冠状面为中心(尽量通过血肿中心)切分脑组织为前后两部分,向前向后取脑冠状脑片各2片,在4片脑片中取包含血肿较小者固定,石蜡包埋,用于HE染色和TUNEL试验;将剩余3片脑片置已预热到37℃的2%TTC溶液中,37℃恒温箱内避光染色20 min,再浸泡在4%多聚甲醛中固定8~12 h。扫描仪扫描标本切面(光学分辨率和色彩位数分别设置为600 dpi,24位真彩色)。观察灶周染色情况;运行图像处理软件运用Visual Basic 6.0,打开扫描图像,测量梗死带内径,单位为mm;测量血肿面积,单位为mm2。对照组同时同部位做相同处理。
1.5 水肿程度评估
取1.4中所述模型的剩余脑组织用电子天平先称取湿重,电热恒温培养干燥两用箱100℃烘烤48 h,再称取干重。利用脑组织含水量=(鲜质量-干质量)/鲜质量×100%进行计算。
1.6 TUNEL法检测凋亡细胞
①标本切片常规脱蜡(不过H2O2),用蒸馏水洗2次,PBS溶液洗2次,每次5 min。②配制新鲜的蛋白酶K(蛋白酶K 20μg溶于10 mM Tris/Hcl中),标本片加新鲜稀释蛋白酶K后37℃烤箱中孵育20 min。③20 min后,取出标本,用PBS洗3次,每次5 min。④闭光环境下加TUNEL反应混合物,加完后在37℃烤箱中孵育1 h,1 h后PBS洗3次,每次5 min。⑤避光环境下加POD 37℃温箱中孵育30 min,后PBS洗3次,每次5 min。⑥DAB显色:取1 ml蒸馏水,依次加入显色试剂盒中A、B、C试剂各一滴,混匀后加至标本片上,镜下控制显色时间3~10 min,水洗。⑦苏木素复染、脱水、透明、封片。
1.7 统计学处理
数据以均数±标准差(x±s)表示。运用SPSS 17.0软件对数据进行统计学分析,相同时间点手术组与对照组的比较采用两样本t检验;手术组不同时间点的组间比较采用单因素方差分析。P<0.05表示差异有统计学意义。
2 结果
2.1 HE染色
光镜下可见出血灶与远隔正常脑组织间有一过渡带即血肿周围区。3 d时,红细胞溶解,出血灶周炎细胞增多以分叶核中性粒为主,水肿明显,出现少量红色的坏死神经元;5 d时出现少量红细胞溶解后的棕黄色颗粒,炎症细胞,细胞水肿程度与3 d时相似,能看见少量坏死神经元;7 d时依然可见水肿、炎细胞,且水肿细胞间夹杂有核固缩或碎裂的细胞,坏死神经元细胞增加;14 d时水肿细胞明显减少,大量坏死细胞及细胞框架,炎细胞及神经元结构不好辨认;21 d时血肿吸收者留有中风囊,囊周细胞基本正常。血肿未吸收者周边丧失细胞结构,色淡染,只见不全细胞结构和坏死后的轮廓。
2.2 TTC染色
正常脑片被均匀染成玫红色,灶周区域色淡染,梗死组织呈基本规则的白色带条状,血肿呈灰黑色。梗死带结果见表1,血肿大小变化结果见表2。
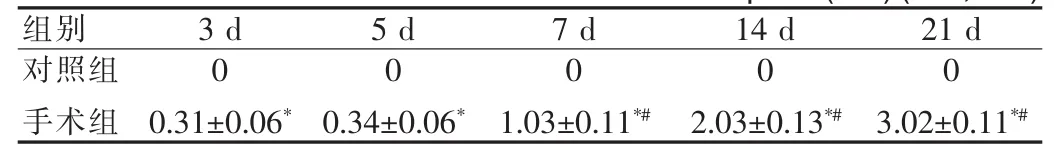
表1 血肿周围梗死带最小内径(mm)(x±s,n=9)Tab.1 Minimum inside diameteraround infarctionspace(mm)(x±s,n=9)
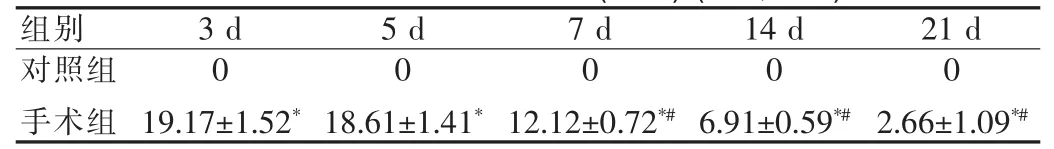
表 2 血肿面积(mm2)(x±s,n=9)Tab.2 Haematoma area(mm2)(x±s,n=9)
2.3 水肿程度评估
脑组织含水量=(鲜质量-干质量)/鲜质量×100%。见表3。
2.4 凋亡细胞检测
阳性细胞为细胞核呈棕黄色,较小的棕黄色圆形小体为凋亡小体。在10×40倍光学显微镜下,围绕梗死灶、出血灶中心取6个不重复高倍镜视野,计数每个视野凋亡细胞阳性细胞数,取均值。见表4。
3 讨论
近年来的临床和动物实验结果表明脑出血神经损伤机制除血肿引起的急性神经组织损害外,还存在血肿周边的继发性损害。有研究显示,急性脑出血灶周存在血流量下降(CBF),且急性期血肿周边灌注缺损量与血肿体积呈正相关,但脑出血后4~6 h血液开始凝固缩小,血肿的机械压迫作用造成的CBF下降在程度和时间上能否达到引起脑缺血性损伤的水平目前仍有争议[7-8]。目前文献报道的相关试验大多集中于脑出血急性期灶周改变,3 d后的血肿灶周改变报道较少。Daverat等[9]的一个前瞻性研究发现,年龄是人类脑出血后功能恢复的重要因素之一;另有研究发现年老的大鼠制作为脑出血模型后,可诱导出相对于年轻大鼠更高的神经胶质细胞活性[10],提示迟发型脑水肿的出现几率可能与年龄成反比(年龄越大,出现几率越小)。本实验模型在成年大鼠(200 g左右)脑内注入自体血100μl,相当于年轻人基底节区脑出血80 ml左右,这一血量可使血肿在3周内不会完全被吸收,且把年龄影响因素降到最小。本试验结果显示:水肿在3~7 d一直处于水肿高峰,14 d才明显下降,与之前的研究结果3~5 d水肿达高峰时间延长,可能因为出血量大,吸收减慢,延长了各种水肿因素的作用时间。同时还显示,脑出血血肿与出血灶远隔区域存在一个区域。随着出血时间延长,此区域细胞形态改变、炎症、组织水肿,组织缺血经历了一个从轻到重的动态改变过程,任其发展,该区则发生不可逆损伤:出现与缺血梗死相似的病理变化。

表3 各时间点大鼠脑组织含水量的变化(%,n=9)Tab.3 The changing of rats brain water content in different time spot(%,n=9)

表4 各时间点凋亡细胞阳性表达(x±s,n=9)Tab.4 The counting of apoptosis in different time spot(x±s,n=9)
脑出血引起细胞凋亡的详细机制还不明确。出血周围继发性缺血、凝血酶释放、血红蛋白分解、炎症细胞浸润、各种细胞因子的表达等因素可能都是诱导脑出血血肿灶周细胞凋亡的因素。有研究显示脑出血导致血肿周围及远隔区域脑组织血流量下降,一般可降至正常的50%左右[11],相对温和的缺血不足以引起脑梗死,但易导致凋亡性细胞死亡[12]。本实验中各手术组间凋亡细胞数均有显著差异,HE染色提示随时间的延长出现不可逆的神经细胞坏死,可能是由于血流量进一步下降,加上凝血酶、血红蛋白释放及各种炎症因子表达增加等因素,导致出血灶周凋亡细胞表达持续增加,最终发展为细胞坏死,血管闭塞,组织血液供应终止,缺血性损伤发生。通过该实验我们认为脑出血后出血灶周为一水肿缺血混合区域,从出血开始到出血后2周以水肿为主,2周后水肿相对稳定,出现缺血改变,类梗死过程;细胞死亡形式开始以凋亡为主,后发展为细胞坏死,所以试验中14 d后,相似位置观察到的凋亡细胞明显下降,出现大量坏死细胞结构。
本实验对于脑出血水肿高峰期后的病程变化有一个较为直观的阐述,为脑出血的治疗,特别是在脱水治疗时间方面提供新的思路。此外,通过试验我们可以看到细胞凋亡在脑出血后是极为重要的一个环节。因此,加大细胞凋亡在出血性脑损伤中的作用及脑出血导致细胞凋亡的原理,利用各种方法阻止细胞凋亡的启动和发展,有可能成为防治出血性卒中的新途径。
[1]Heo JH,Han SW,Lee SK.Free radicals as triggers of brainedema formation after stroke[J].Free Radie Biol Med,2005,39(1):51-70.
[2]Kimelberg HK.Current eoncepts of brainedema:review of laboratory investigations[J].JNeurosurg,1995,83(6):1051-1059.
[3]Forbes KP,Pipe JG,Heiserman JE.Diffusion-weighted imaging provides support for secondary neuronal damage from intraparenchymal hematoma[J].Neuroradiology,2003,45:363-367.
[4]Deinsberger W,Vogel J,Kuschinsky W,et al.Experimental intracerebral hemorrhage:description of a double injection model in rats[J].Neurol Res,1996,18(5):475-4777.
[5]周中和,曲方.一种改良大鼠自体血脑出血模型:二次注血/退针法[J].中国临床神经科学,2004,12(4):406-408.
[6]Bederson JB,Pitts LH,Tsuji M,et al.Rat middle cerebral artery occlusion:evaluation of the model and development of a neurologic examination[J].Stroke,1986,17(3):472-6.
[7]Xue M,Del-Bigio MR.Intracortical hemorrhage injury in rats:relationship between blood fractions and brain cell death[J].Stroke,2000,31(7):1721-1727.
[8]Peeling J,Yan HJ,Corbett D,et al.Effect of FK-506 on inflammation and behavioral outcome following intracerebral hemorrhage in rat[J].Exp Neurol,2001,167(2):341-347.
[9]Daverat P,Castel JP,Dartigues JF,et al.Death and functional outcome after spontaneous intracerebral hemorrhage:a prospective study of 166 cases using multivariate analysis[J].Stroke,1991,22(1):1-6.
[10]Gong Y,Hua Y,Keep RF,et al.hemorrhage:elects of agingon brain edema and neurological deficits[J].Stroke,2004,35(11):2571-2575.
[11]Yonezawa T,Hashimoto H,Sakaki T.Serial change of cerebral blood flow aftercollagenase induced intracerebral hemorrhage in rats[J].No Shinkei Geka,1999,27(5):419-425.
[12]Bennett SA,Tenniswood M,Chen JH,et al.Chronic cerebral hypoperfusion elicits neuronal apoptosisand behavioral imparement[J].Neuroreport,1998,9(1):161-166.
猜你喜欢
中国生殖健康(2020年8期)2021-01-18
基层中医药(2020年4期)2020-02-13
现代临床医学(2019年6期)2019-12-07
中国生殖健康(2018年3期)2018-11-06
中国组织化学与细胞化学杂志(2017年1期)2017-06-15
中成药(2017年6期)2017-06-13
临床医药文献杂志(电子版)(2017年11期)2017-05-17
Coco薇(2016年8期)2016-10-09
吉林大学学报(医学版)(2015年4期)2015-12-17
癌变·畸变·突变(2015年3期)2015-02-27
