
固相萃取-HPLC法测定白果中总银杏酸的含量Δ
2011-05-21 05:39:50鲍家科宋晓宁张志勇张志宇吴克楠贵州省药品检验所贵阳市550004
中国药房 2011年27期
罗 曼,鲍家科,宋晓宁,张志勇,张志宇,吴克楠(贵州省药品检验所,贵阳市 550004)
白果为银杏科植物银杏Ginkgo biloba L.的干燥成熟种子,秋季种子成熟时采收,除去肉质外种皮,洗净,稍蒸或略煮后,烘干[1]。白果具有多种生物活性物质,银杏酸是其毒性成分,具有致敏性、胎盘毒性和免疫毒性作用[2~4],对人类健康有潜在危害,但现行质量标准中尚未控制白果中银杏酸的含量。因此,应建立准确的分析方法,对白果及其炮制品中的银杏酸进行控制,为白果及其炮制品的研究提供理论基础。
固相萃取(solid-phase extraction,SPE)是近年发展起来的一种样品预处理技术,由液-固萃取和柱液相色谱技术相结合发展而来,由于具萃取效率高、选择性好、适用范围广、操作简单、省时等优点,目前已被较为广泛地应用于环境分析、食品分析、生物样本中毒物的分析[5,6]中,在复杂中药有效成分分析方面的应用也日益增加[7~9]。银杏酸在白果中含量甚微,药材中又存在大量复杂基质,用传统的提取分离方法较难有效地从白果中提取银杏酸。笔者参考2010年版《中国药典》(一部)银杏叶提取物总银杏酸检查项下方法[1]及有关文献[10],采用C18固相萃取小柱对样品进行纯化精制,拟定白果中总银杏酸的含量测定方法,其准确度与重复性考察均获得了较满意的结果,适用于白果中总银杏酸的含量分析。
1 仪器与试药
Agilent 1100型高效液相色谱(HPLC)仪、色谱工作站(美国Agilent公司)。
总银杏酸对照品(中国药品生物制品检定所,批号:110770-220510);甲醇为色谱纯,其余试剂均为分析纯;8批市售白果药材分别来源于贵州贵阳、贵州罗甸、贵州水城、四川都江堰、四川汶川等地,经本所熊慧林主任药师鉴定为银杏科植物银杏G.biloba L.的干燥成熟种子。
2 方法与结果
2.1 色谱条件
色谱柱:十八烷基硅烷键合硅胶柱(150mm×4.6mm,5µm);流动相:甲醇-3%冰醋酸(91∶9);检测波长:310 nm;柱温:30 ℃;流速:1.0mL·min-1。
2.2 对照品溶液的制备
精密称取总银杏酸对照品20.73mg,置100mL量瓶中,加甲醇溶解并稀释至刻度,摇匀,得对照品贮备液(0.2073mg·mL-1)。再精密量取5mL此贮备液,置10mL量瓶中,加甲醇溶解并稀释至刻度,即得对照品溶液(0.10365mg·mL-1)。
2.3 供试品溶液的制备
取白果细粉(过三号筛)约2.5 g,置具塞锥形瓶中,精密称定,精密加入无水甲醇25mL,称定,80℃水浴中回流3 h,放冷,用无水甲醇补足减失的重量,摇匀,滤过,精密量取续滤液15mL,80℃水浴挥至近干,用75%甲醇2mL溶解转移至已处理好的C18固相萃取小柱(先用无水甲醇10mL冲洗,再用75%的甲醇溶液10mL冲洗,小柱上加脱脂棉少许)上,继续用75%甲醇5mL洗脱,洗脱液弃去。再用无水甲醇约4.5mL洗脱,洗脱液收集于5mL量瓶中,加无水甲醇稀释至刻度,摇匀,用0.45 μm微孔滤膜滤过,即得。
分别精密吸取对照品溶液10µL与供试品溶液10~20µL,注入液相色谱仪测定,按外标法计算,即得。
2.4 专属性考察
在“2.1”项下色谱条件下,总银杏酸与其他组分可达到基线分离。色谱见图1。
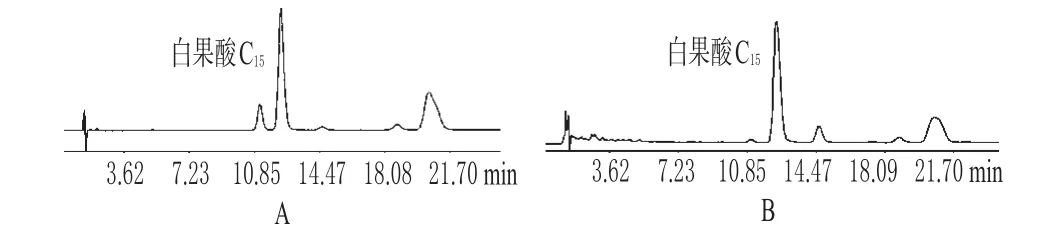
图1 高效液相色谱图A.总银杏酸对照品;B.供试品Fig 1 HPLC chromatogramsA.ginkgolic acids control;B.test sample
2.5 线性关系考察
分别精密吸取总银杏酸对照品溶液(0.10365mg·mL-1)6、10、14、20、24 µL(0.6219、1.0365、1.4511、2.0730、2.4876μg),注入液相色谱仪测定,按“2.1”项下色谱条件测定总峰面积。以总峰面积积分值(Y)为纵坐标,总银杏酸进样量(X)为横坐标,制备标准曲线,得回归方程为Y=413.2750X-4.2990(r=0.9999,n=5)。结果表明,总银杏酸进样量在0.6219~2.4876μg范围内与总峰面积积分值呈良好线性关系。拟合成过原点的直线方程为Y=410.9284451X。将线性关系考察的最低进样量与最高进样量分别代入两方程进行计算,所得峰面积相对偏差为0.62%(低)~0.0031%(高),由此可作截距近似为0,故本文中采用外标一点法计算总银杏酸含量。
2.6 精密度试验
精密吸取总银杏酸对照品溶液(0.10365mg·mL-1)10 µL,注入液相色谱仪,重复测定5次,按“2.1”项下色谱条件测定总峰面积。结果,RSD=1.08%(n=5),表明仪器精密度良好。
2.7 重复性试验
取白果细粉(过三号筛)约2.5 g,置具塞锥形瓶中,精密称定,共6份,分别按“2.3”项下方法制备供试品溶液,精密吸取20μL,进样测定含量。结果,总银杏酸的平均含量为0.2021mg·g-1,RSD=1.58(n=6),表明本方法重复性良好。
2.8 稳定性试验
取白果细粉(过三号筛)约2.5 g,精密称定,置具塞锥形瓶中,按“2.3”项下方法制备供试品溶液,分别于不同时间(0、2、4、6、8、10、12 h)精密吸取 20μL,进样测定。结果,RSD=0.74%(n=7),表明供试品溶液在12 h内稳定性良好。
2.9 加样回收率试验
取已测知含量(0.2021mg·g-1)的样品约1.3 g,精密称定,共6份,置具塞锥形瓶中,分别精密加入对照品溶液(0.2073mg·mL-1)1.3mL,置80 ℃水浴上挥尽溶剂,放冷,自“精密加入甲醇25mL,称定……”起,按“2.3”项下方法操作,计算加样回收率,结果见表1。
2.10 耐用性试验
选择3种不同品牌色谱柱[Aglient Ecilipse XDB-C18(150mm×4.6mm,5µm)、Waters Symmetry C18(150mm×4.6mm,5µm)、Ultimate XB-C18(250mm×4.6mm,5µm)],检测同一批次

表1 加样回收率试验结果(n=6)Tab 1 Results of recovery tests(n=6)
2.11 样品含量测定
取市售白果药材8批,依法测定含量,结果见表2。样品,每一批次平行测定2份。结果,RSD=1.93%(n=6),表明色谱柱的改变对测定结果无显著影响。
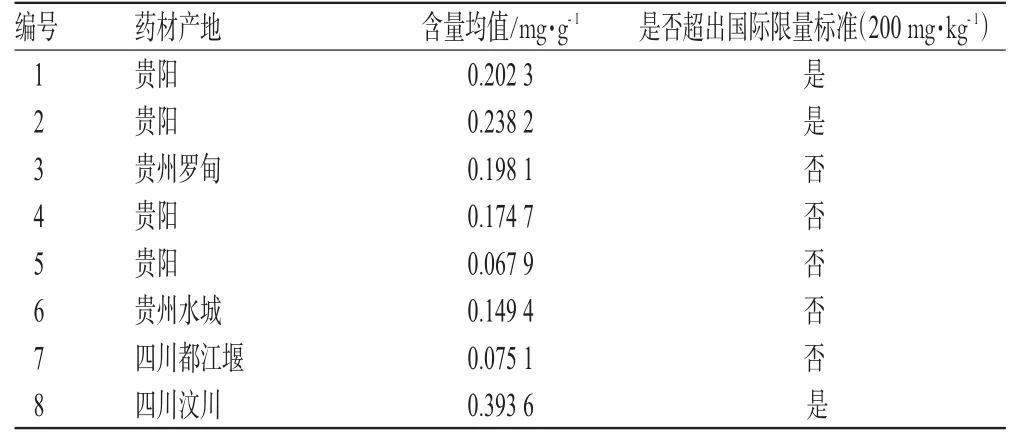
表2 样品含量测定结果Tab 2 Results of content determination of samples
3 讨论
白果既含有大量水溶性杂质,又含有较多的亲脂性杂质。由于总银杏酸有一定的脂溶性,笔者曾采用正己烷、石油醚等极性较低的溶剂提取总银杏酸,硅胶柱精制供试品,但仅有微量银杏酸检出,表明方法不适用于白果中银杏酸的分析。又曾参考文献以极性较低溶剂石油醚索氏提取样品,以去除白果中大量极性较大的水溶性杂质,但经试验发现石油醚的提取效率不高,平行样品测定结果的偏差较大。考虑到银杏酸在甲醇中易溶解,本研究采用80℃甲醇回流提取样品,同时考察了提取时间,以回流3 h的测定值最高,若增加提取时间,含量测定值不再增高,从而确定样品提取方法为80℃甲醇回流提取3 h。但甲醇提取物含杂质较多,会干扰测定,且损害色谱柱的使用寿命。故本研究采用与液相色谱柱填料相近的C18小柱进行固相萃取,甲醇提取物以75%甲醇溶液洗脱,除去极性较大的杂质,再收集一定体积的甲醇洗脱液,洗脱液中含总银杏酸,剩余的大量脂性杂质则保留在小柱上,从而有效地除去了杂质的干扰,保证了测定结果的准确性。
笔者考察了不同体积分数的甲醇溶液作洗脱溶剂的效果。结果发现,使用75%甲醇溶液作洗脱剂,收集量为5mL,洗脱液中检出大量杂质但无总银杏酸;再收集5mL洗脱液,洗脱液中仅有少量杂质检出。再以80%甲醇溶液5mL作洗脱剂,洗脱液中有少量总银杏酸检出。因此,确定采用75%甲醇溶液5mL作洗脱剂,可洗脱试样中大量的极性较大杂质,保留于小柱上的总银杏酸再以约5mL无水甲醇洗脱,获得了较满意的结果。
试验比较了3种不同品牌色谱柱(见“2.10”项),在上述色谱条件下,样品色谱中总银杏酸各峰与其他杂质峰均能达到基线分离。3种色谱柱的柱效以总银杏酸组峰中白果酸C15峰计算均不低于5000,考虑到色谱柱的差异及使用寿命等影响因素,暂定在分离度符合药典要求的前提下,色谱柱的理论板数按白果酸C15峰计算应不低于3000。
经试验,发现有部分批次市售白果的总银杏酸含量超过国际限量标准(200mg·kg-1),且不同来源渠道白果中银杏酸含量差异较大,这可能与银杏产地、采收时间、银杏树龄及药材采收后的处理条件等有关。试验结果提示应对白果的食用安全性引起重视,增加总银杏酸的质控指标,并对药材采集后的处理条件参数(如蒸、煮、烘干等环节中的具体温度、时间、压力等)作进一步的明确。
[1]国家药典委员会编.中华人民共和国药典(一部)[S].2010年版.北京:中国医药科技出版社,2010:100.
[2]Jaggy H,Koch E.Chemistry and biology of alkyphenols from Ginkgo biloba L.[J].Pharmazie,1997,52(10):735.
[3]Baron-Ruppert G,Luepke NP.Evidence for toxic effects of alkyphenols from Ginkgo biloba in the hen’s egg test[J].Phytomedicine,2001,8(2):133.
[4]Koch E,Jaggy H,Chatterjee SS.Evidence for immunotoxic effects of crude Ginkgo biloba L.leaf extracts using the popliteal lymphnode assay in the mouse[J].Int J Immunopharmacol,2000,22(3):229.
[5]郑仁达,洪 冰.固相萃取反相高效液相色谱法测定人血浆中兰索拉唑的浓度[J].中国药房,2010,21(30):2834.
[6]王庆芬,郑绍忠,张铭穷,等.复方甘草口服溶液中吗啡含量测定方法的验证[J].中国药房,2008,19(36):2839.
[7]张 锐,彭杨锐.SPE-HPLC测定防芷鼻炎胶囊中升麻素的含量[J].中成药,2007,29(12):1865.
[8]徐 活,黄京芳,陈 平.固相萃取-高效液相色谱法测定儿泻止颗粒中葛根索含量[J].中国医院药学杂志,2004,24(6):337.
[9]陈 蕾,李永庆,朱霁虹,等.SPE-HPLC法测定菊延保康颗粒中延胡索乙素的含量[J].中成药,2003,25(8):629.
[10]仰榴青,吴向阳,陈 均.HPLC法测定白果中银杏酸的含量[J].药物分析杂志,2004,24(6):636.
猜你喜欢
儿童时代·快乐苗苗(2022年10期)2022-12-09 08:54:04
时代邮刊·下半月(2021年10期)2021-10-23 04:35:24
幼儿教育·父母孩子版(2021年6期)2021-08-05 05:48:40
艺术品鉴(2020年6期)2020-12-06 10:49:08
幼儿教育·父母孩子版(2020年8期)2020-03-04 11:54:24
基层中医药(2018年11期)2019-01-31 05:27:04
现代园艺(2018年2期)2018-03-15 08:00:06
领导文萃(2017年6期)2017-03-24 09:31:39
中学生数理化·高一版(2016年7期)2016-12-07 20:47:07
饮食科学(2016年3期)2016-07-04 08:35:04
