
原料药无菌工艺模拟验证国际认证要求
2011-02-27 02:43高海燕陈军丽丁恩峰
化工与医药工程 2011年2期
高海燕 陈军丽 丁恩峰
(1. 石药集团恩必普药业有限公司,石家庄 050031;2. 石家庄市第三医院制剂科,石家庄 050031;3. 英国施达化学集团公司中国代表处,石家庄 050031)
在欧美药政法规体系里面,如果无菌制剂采用无菌原料药(API)来制备,那么,无菌原料药的无菌性质对于制剂来说是至关重要的。无菌原料药一般采用两种方法来制造:灭菌工艺和无菌工艺。当原料药采用无菌工艺制造时,因为这种工艺无菌保证水平(SAL)较低,因此,需要采用模拟验证来评估无菌工艺的保证能力。在目前制药行业,通常采用无菌工艺模拟验证(也称为培养基灌装)方式评估工艺无菌保证能力。
目前制药行业对于无菌制剂的工艺模拟验证研究较多,而对于无菌原料药的工艺模拟验证研究较少。例如,工艺模拟验证实施细节问题、最后判定标准选择问题等,都存在很多模糊或者错误的认识。
1 各国GMP法规指南对无菌工艺验证的要求
对于原料药的管理,中国药政法规和欧美药政法规是不同的。在欧美国家,原料药不被作为药品来管理,而是作为活性物质(API)来管理。因为欧美药政法规对于原料药的无菌工艺模拟验证没有很明确的要求,目前法规和指南中可以找到关于无菌工艺模拟验证的要求,主要适用于无菌制剂的工艺模拟验证。中国药政法规由于建设较晚,还没有很完善的体系,对于无菌原料药工艺模拟验证,也没有针对性的要求。下面列表给出了各国对于无菌工艺模拟药政的要求。
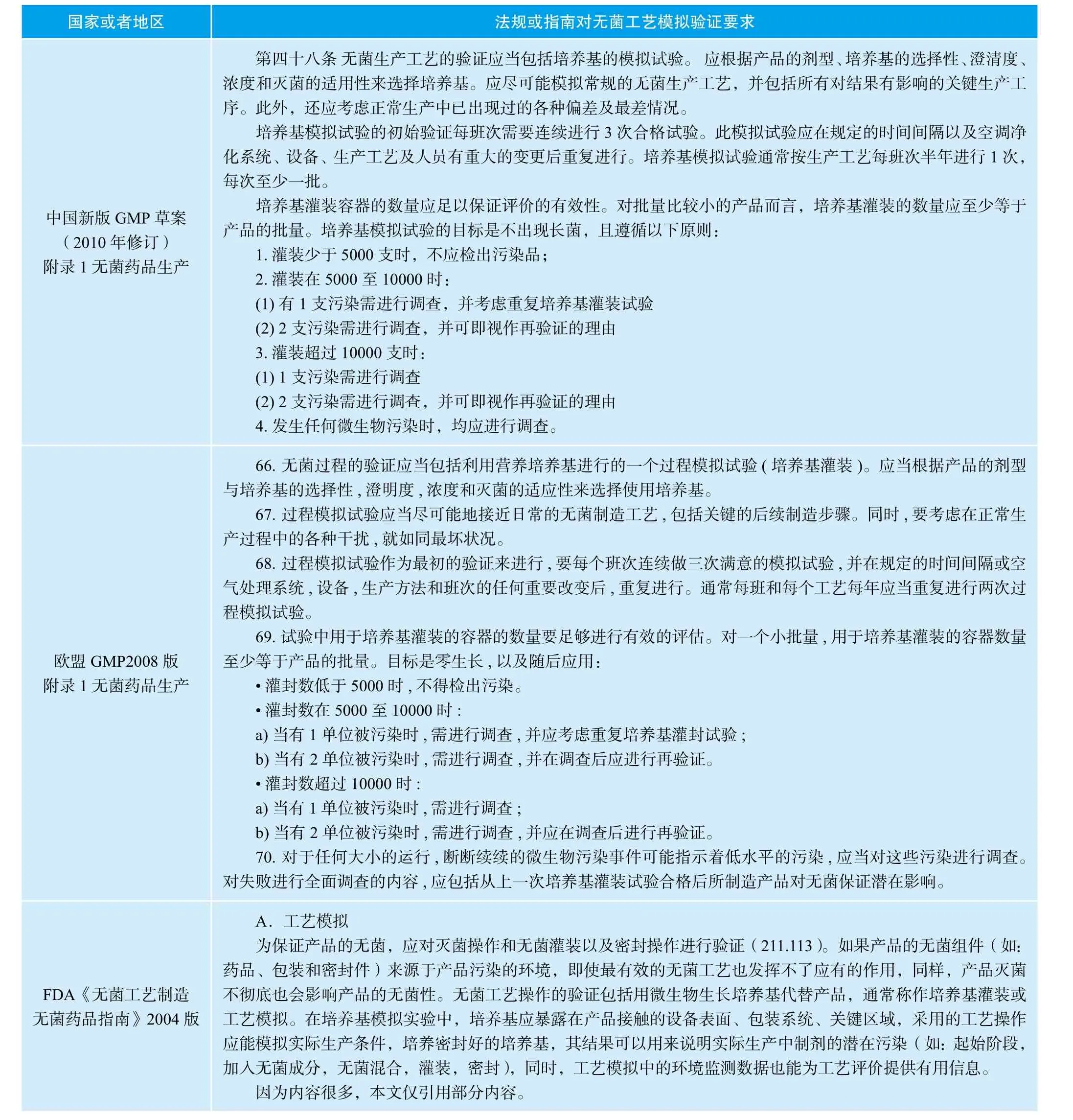
表1 各国GMP法规指南对无菌工艺验证的要求
2 无菌原料药工艺模拟验证介绍
2.1 无菌原料药工艺模拟验证方法学问题
在进行无菌原料药工艺模拟验证之前,验证人员必须全面深刻地评估无菌原料药制造工艺,并确定无菌工艺模拟验证的范围,这是无菌工艺模拟验证成功的关键。
由于无菌原料药生产工艺一般包括多个步骤,模拟验证可以采取2种方式:第一种方式:分别评估每部操作的无菌保证能力;第二种方式:综合评估无菌操作步骤组合的无菌保证能力。下面介绍这2种方式。
2.1.1 一次性全部工艺模拟方法(Total Process Simulation)
分析比较,可以得到第一种方式的优点和缺点如下:
优点:
— 这种组合模拟方法比那些分段模拟方法更接近实际工艺;
— 比逐个单元模拟节省验证时间;
缺点:
— 如果发现污染样品,监测和确定污染来源很困难;
— 如果试验失败,在纠正措施制定以后,必须进行全部步骤的再次模拟验证,成本很高;
— 在整个模拟过程中,一般要求使用单一的测试物料(全是液体物料或者全是固体物料),这可能引入和实际工艺显著不同的操作步骤。并且污染如何产生的情况,也可能和实际工艺差距甚大。
2.1.2 单元操作模拟方法(Unit Operation Simulations)
通过分析和比较,可以得到第二种方式的优点和缺点如下:
优点:
— 如果发现污染样品,可以把来源限制在更小的范围来调查;
— 对特定步骤变更效果的评估可以限制在较小的范围;
— 如果某个环节失败,在确定纠正措施以后,只需要重复那个失败的环节,这样做较节约成本;
— 在某些工艺中,这些单元操作的组合,可能更接近实际工艺;
— 在不同的步骤可以使用不同的物料(固体或者液体),这可能更接近实际情况。
缺点:
— 比一次性模拟验证浪费时间;
— 可能要求不同程度的重复工作来评估全部工艺;
— 评估单个单元操作的方法可能要求对无菌物料更多的处理来满足一个单独工艺模拟测试;
— 在每个单元模拟试验中产生大量的环境监控样品,需要处理,增加验证成本。
2.2 无菌工艺模拟验证的介质选择问题
2.2.1 模拟验证介质选择原则
根据PDA技术报告 No.28《Process Simulation Testing for Sterile Bulk Pharmaceutical Chemicals》,模拟介质选择原则有两个。第一个原则是,应该选择促微生物生长培养基、或者安慰剂、或者实际产品(一般选择辅料)、或者不使用任何介质。另外,还要确定选用液体介质还是固体介质,例如:在结晶工艺中,前段工艺需要选择液体介质模拟,后段工艺需要选择固体介质来模拟验证。第二个原则是,要考虑模拟介质的安全性问题;所选用模拟介质不能造成不可消除的潜在污染。
下面列表介绍4种模拟介质选择方式和优缺点。

表2 4种模拟介质选择方式及优缺点
2.2.2 模拟介质选择的具体经验
— 直接选用无菌的安慰剂可能会节约时间;
— 选择的模拟物料不能影响无菌检测;
— 根据设备和操作工艺特点,可能选择液体物料或者固体物料;
— 可以选择促微生物生长物料或者惰性物料;
— 在某些工艺模拟中,可能会使用多种物料;
— 不论什么模拟物料,最后包装要接近真实产品。
2.2.3 选择粉末状介质的经验
— 无菌干粉促生长培养很少被选择,因为,流动性很差,通过无菌处理设备很难;
— 根据制药行业经验,主要选用lactose, mannitol和polyethylene glycol;
— 干粉培养基最常用dry Soybean-Casein Digest Medium (SCDM)大豆消化酪素琼脂培养基 ;
— 被选择物料必须容易灭菌、溶解和分散,不抑制微生物的生长,而且容易在设备里面被处理。
2.2.4 选择液体模拟介质的经验
— 促生长培养基容易溶解,可以被选用;但是使用后的清洁问题需要关注;
— 可以选用的液体培养基:SCDM(TSB)和peptone broth(蛋白胨肉汤培养基);— 如果不影响促生长能力,培养基可以被稀释;— WFI和其他溶媒可以作为模拟流体,但是溶媒不能具有抗微生物活性;
— 液体物料一般通过0.22 μm无菌过滤系统来获得无菌性质。
2.3 无菌工艺模拟验证的前提条件
2.3.1 生产设施和设备验证情况
尽管无菌工艺模拟验证不是正式的工艺验证,但是,模拟验证也是针对无菌工艺本身的一次较全面的评估和核实。因此,在进行无菌工艺模拟验证之前,用于生产无菌原料药的各种设备和系统,应该先验证并符合相关要求。
2.3.2 公用系统验证情况
用于生产无菌原料药的公用系统,例如:HVAC系统、注射用水系统、纯化水系统、压缩空气系统、真空系统、蒸汽灭菌系统,以及用于空间消毒的臭氧消毒系统等,都应该在无菌工艺模拟验证之前,先进行验证并符合要求。
2.3.3 无菌检验方法验证情况
为了准确评估无菌工艺的保证能力,正确检测和评估样品的无菌性质,用于无菌检验的方法应该先经过验证并符合相关要求。
2.3.4 无菌过滤工艺验证情况
采用无菌工艺制备的无菌原料药,多是对热敏感的物料,不能耐受热力灭菌方法。在这种情况,无菌过滤系统是将非无菌物料转换为无菌物料的关键系统,应该在无菌工艺模拟验证之前先进行验证。无菌过滤系统的验证,除了需要进行滤芯和过滤药液相容性研究以外,还应该研究不同过滤时间的影响、完整性测试项目和挑战性试验。
2.3.5 相关支持灭菌工艺验证情况
一种无菌原料药生产过程中,会使用结晶罐、溶媒罐、各种流体管道和气体管道、还可能会用到各种器具以及操作人员穿戴的洁净服。这些设备和器具在生产前都应该进行灭菌,并应该保存在规定条件下。这些支持系统,在无菌工艺模拟验证之前,都应该先验证完毕并符合要求。
例如:洁净服的灭菌,需要考察灭菌条件的有效性,还需要考察保留时间(holding time)的长短。保留时间是无菌工艺模拟验证中涉及的一个干扰项目,应该给与重视。
2.3.6 清洁规程验证情况
无菌工艺模拟验证中,很多情况下不是使用真实物料进行工艺模拟,而是使用促微生物生长培养基或者安慰剂进行工艺模拟。在这种情况下,原来起草的设备或者器具的清洁规程就失去了针对性,应该根据新的情况起草有针对性的清洁规程。
这些专门为无菌工艺模拟验证起草的清洁规程的验证安排是一个问题:如果提前进行清洁验证,会浪费很多物料;如果和无菌工艺模拟验证同时进行,一旦出现验证失败情况,会很难确定具体原因。因此,在清洁验证和无菌工艺模拟验证的安排中,应该基于实际情况和风险分析结果,做出更具有针对性的安排。
2.3.7 人员培训和人员健康情况
所有参加无菌工艺模拟验证的人员,除了要接受相应岗位规程和知识的培训,都要统一参加无菌工艺模拟验证方案的培训。培训完毕,需要进行考核;符合要求者可以参加无菌工艺模拟验证工作。
另外,针对参加验证人员的健康检查,也应该根据验证情况来核实最近一次人员的体检情况。
2.3.8 验证文件准备情况
除了按照下面第4部分准备验证方案和验证报告以外,无菌工艺模拟验证需要的各种表格、记录和台帐都应该准备完善,和正式工艺验证没有很大区别。
2.3.9 模拟生产批记录准备情况
模拟生产批记录是无菌工艺模拟验证的关键性文件,需要由生产部门技术人员单独起草。在模拟验证过程中,由于模拟物料的引入,导致很多例行的工艺环节被改变或者被省略,另外,一些新的工艺环节被加入,因此,需要起草一份新的专门针对模拟生产的批记录。
由于模拟生产的产品都是用于培养,不会被包装和销售,因此,包装记录一般不需要准备。
模拟生产样品的检验记录需要根据实际情况专门准备。
2.4 无菌工艺模拟验证的文件管理问题
2.4.1 验证方案包含的信息
在无菌原料药工艺模拟验证之前,需要起草和审核并批准工艺验证方案。工艺验证方案应该包括如下信息:模拟验证的工艺描述、生产进行的区域描述、需要评估的灌装线和设备介绍、容器和密封件类型、灌装速度选择原因、灌装样品数量、模拟验证中要包括的干扰和中断的数目和类型、参加人员数目、需要使用的培养基或者其他模拟介质、灌装体积、培养条件(例如:培养时间和温度等)、需要进行的环境监测计划和规程、批生产记录的复印件一份、各个测试项目的接受标准、选择最差条件的原理和考虑点、阳性样品所处位置描述、促生长试验结果、其他要素等。
2.4.2 验证报告包含信息
在验证结束后,应该由验证小组负责人或者指定人员撰写验证报告,内容应该包括如下信息:批生产记录是验证报告的重要组成部分,要如实、及时记录各项操作,例如:清洁情况、灭菌情况、样品传递和培养等;对于采用的干扰条件,要详细记录干扰的数量和类型;灌装数量、培养数量、阳性样品数量、拒绝的样品数量(破损容器等)、促生长试验结果、工艺无菌保证能力评估的结论。
2.5 无菌工艺模拟验证的实施细节问题
2.5.1 干预情况
为了更好的评估无菌工艺保证能力,在无菌工艺模拟验证过程中,可以设定一些干预措施,更好的评估和研究无菌工艺。建议如下:
— 模拟验证必须包括日常操作活动,例如:设备调整、容器密封件输送供应和取样等活动;
— 非日常活动(突发干预行为)可以加入模拟验证;
— 在模拟验证中,非预期的干预活动是可以加入的,例如:液体泄漏、设备停止再启动等;
— 如果最后模拟验证顺利通过,这些非预期干预可以作为日常活动的一部分。
2.5.2 持续时间
根据制药企业的经验,产品暴露时间和无菌合格率具有一定的关系。在设计无菌工艺模拟验证方案时,应该考虑如下要求:
— 必须保证足够工艺持续时间来展示无菌工艺的保证能力;
— 最初的准备活动、设备调整、手动维护等应该加入模拟验证中;
— 模拟验证还应该包括日常的,或者非典型的干预活动;
— 作为正常操作的一部分,更衣、中间休息和换班都应该包括在模拟验证中;
— 原料药生产中和污染相关性最大的环节是磨粉和过筛,这些重复工作长期进行,人员对于产品污染影响最大,应该给与考虑。
2.5.3 批量选择
批量的选择和最后合格标准具有一定关系。如果模拟验证用于支持研发申报工作,模拟验证批量可以减小,保持和研发阶段生产批量一致即可。如果模拟验证工艺属于商业化生产工艺,需要考虑如下要求:
— 模拟验证应该使用足够的物料,确保接触所有可能影响无菌的设备表面;
— 批量选择要满足可以最大程度的模拟实际工艺操作;
— 模拟验证批量和实际批量不必相同。
2.5.4 培养条件
根据PDA TR No.28和其他相关指南,样品的培养一般采取如下条件:
— 最少培养14天,这是最低要求;
— 培养温度每个公司可以采取不同的策略;
— 培养温度应该有利于微生物的复活和恢复;这些微生物应该主要来自环境监控和产品微生物检验中发现的微生物;
— 一般选择20 ~ 35℃来培养;更合适的建议是分段培养,就是在20 ~ 25℃范围培养7天,然后在30 ~ 35℃范围培养7天;
— 培养期间温度应该持续监控。
2.5.5 操作规程
无菌药品的生产,要非常关注细节和人员的熟练程度。为了确保将失误降低到最低水平,应该给操作人员提供高质量的SOP。
2.5.6 人员影响
因为原料药生产中,人工操作环节较多,影响较大;应该确保每个员工在一定周期内至少参加一次成功的无菌工艺模拟验证。如果因为车间人员和班次较多,应该至少保证再验证时,要将未参加模拟验证的人员包括进来。
2.5.7 生产周期
因为原料药生产的特殊性,一些特殊情况需要采用特殊办法来处理。例如:某些公司在密闭系统里面,连续生产多批次原料药而不进行中间的清洁状态,对于这种模式,模拟验证应该证实这种模式在开放系统里面的接受可能。这样做的原因是,任何一种产品生产不能一直处于密闭系统中,在一定周期结束后,会进行一个密闭系统到开放系统的转换,这也是容易染菌的环节,因此,需要关注这个环节。
2.6 无菌工艺模拟验证的判断标准和结果解释
毫无疑问,任何无菌药品的生产都是追求0污染率。但是在实际情况下,由于多种客观条件的限制,这个目标要达到是有难度的。对于原料药,在很多环节不可避免的要引入很多人员操作活动,这些污染几率不可避免。鉴于此,对于无菌原料药工艺模拟验证接受标准是有不同看法的。
PDA TR No.28提供了两种选择,供大家参考选用。
2.6.1 定性接受标准
如果原料药制药企业采用了高度自动化的生产线,而且无菌取样和无菌检验环节的质量保证水平很高,可以将0污染率作为无菌工艺模拟验证的接受标准。在这种情况下,验证结果就是2种:通过或者失败。
2.6.2 定量接受标准
因为影响最终产品无菌性质的环节甚多,所以,大部分制药企业不会选择定性接受标准,而会选择定量接受标准。
定量接受标准建立的基础是:无菌制剂的无菌性质来自无菌原料药的无差别传递。在这种情况下,可以将法规方对于无菌制剂的验证标准折算到无菌原料药的工艺模拟验证中。
根据FDA《sterile drug products produced by aseptic process-current GMP》2004版指南,无菌制剂工艺模拟验证合格标准为1/10000,即:无菌制剂模拟灌装10000个单位,最多允许出现一个阳性样品。我们将这个验证标准折算到无菌原料药工艺模拟验证中,参见下面例子。
例子:假设某原料药最小生产批量是200 kg,用这种原料药生产的无菌制剂最大装量是10 g,模拟验证中使用安慰剂60 kg,所有的模拟验证样品检测后发现2CFU。计算和推论如下:
— 每批次污染水平就是2CFU,表明生产批次也是这个污染水平;
— 用这批原料药生产制剂最小单位数量计算:200000 g / 10 g = 20 000 units/batch;
— 计算CFU/unit最大值:2/20000 = 0.0001 CFU / unit。
结论:因为0.0001CFU/unit和FDA指南推荐的接受标准相同,因此,这种无菌原料药工艺模拟验证符合要求,通过评估。
2.6.3 阳性样品的调查
无菌工艺模拟验证结束后,不管验证最后是否通过,任何污染样品都要进行调查。调查要遵循如下原则和方法:
— 详尽的取样规程和菌种鉴别规程,是调查成功的基础;
— 如果工艺模拟验证显示了污染的可能来源,必须调查;
— 调查记录应该保留;
— 基于调查结果,污染来源可能是明确的或者是不明确的;
— 污染来源可能是单一来源,也可能是多来源组合;
— 如果来源是明确的,应该建立纠正预防措施并形成书面规程;
— 纠正预防措施可能包括重复进行工艺模拟验证;
— 如果原因没有发现,应该像验证新的无菌工艺那样来再验证一遍。
2.7 无菌工艺模拟验证的再验证问题
为了定期评估无菌工艺的保证能力,需要针对工艺或者设备、设施和人员的变化情况,进行定期或者改变后的再验证。
2.7.1 再验证的频率问题
如果发生影响无菌工艺保证能力的因素变化,应该在变更以后尽快进行无菌工艺模拟再验证。例如:原料药分装岗位人员明显变化后,应该进行无菌工艺模拟再验证。如果初次验证结束后,在一段时间内,没有发生影响无菌工艺保证能力的因素变化情况,需要定期进行再验证。再验证频率根据PDA TR No.28的建议,可以每年进行一次。根据欧美官方的期望和经验,每年至少进行2次无菌工艺模拟验证。
2.7.2 再验证的批次问题
如果再验证情况属于没有变更发生的定期再验证,再验证只需要进行一批次模拟生产验证。如果再验证属于变更性再验证,根据变更对象和变更程度,需要进行1-3批次的模拟生产验证。
2.7.3 再验证的批量问题
如果批量在上一次验证结束后没有发生变化,再验证批量可以和上一次验证时的模拟生产批量相同。
2.8 头孢菌素A无菌工艺模拟验证案例介绍
河北省石家庄某制药公司生产无菌原料药-头孢哌酮钠。本文以此品种为案例,介绍逐步实施无菌工艺模拟验证的方法学和流程。
2.8.1 生产工艺分析
河北某制药公司生产的无菌头孢哌酮钠原料药,属于采用无菌工艺制造的典型无菌原料药。主要工艺步骤包括:合成、配液、无菌过滤、结晶、三合一处理、干燥、分装和外包装工序。
在进行原料药无菌工艺模拟验证前,公司组织质量部、生产部、研发部和工程部对工艺进行无菌关键性分析。分析结果见表3:
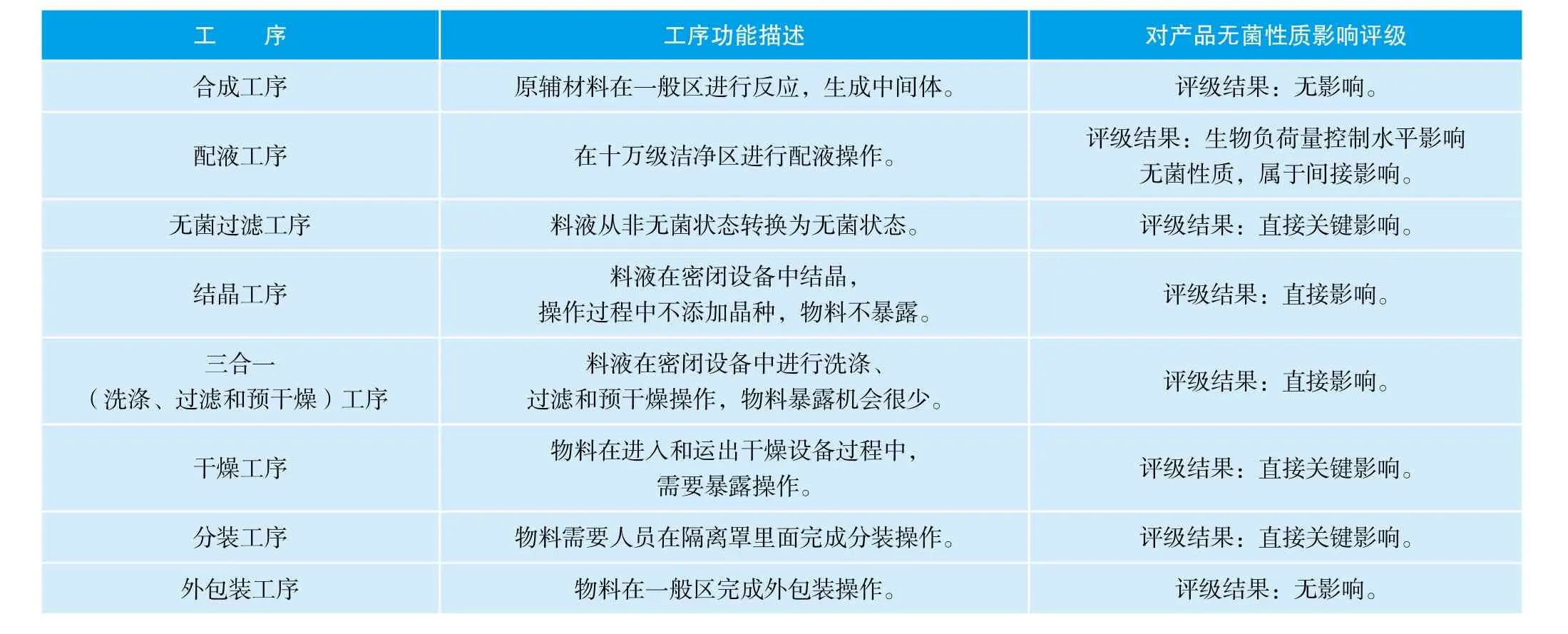
表3 对工艺的无菌关键性分析结果
通过上述工艺分析,确定配液、无菌过滤、结晶、三合一处理、干燥、分装等工序为无菌原料药工艺模拟验证范围,需要在模拟测试中评价相关工序无菌保证能力。
2.8.2 验证准备工作
根据公司关于验证的管理制度,布置相关工作给质量部、生产部、QA、QC、工程部和物料部。这些工作涉及无菌车间HVAC系统和工艺用水验证状态核实、验证包材和培养基选择采购、验证检验试剂的采购等。
2.8.3 文件起草和人员培训工作
由公司生产部负责无菌工艺模拟验证方案的起草,同时,负责模拟验证批记录的起草工作。待草案确定后,按照文件审核规程,由质量部牵头,组成验证小组,专门开会审核验证方案和相关记录。
在验证方案审核结束后,安排要参加模拟验证的人员参加了验证方案和批记录的培训工作。
2.8.4 模拟物料的选择
因为头孢哌酮钠在生产过程中,物料状态存在一个转换过程:固体-液体-固体;因此在配液、结晶和三合一工序采用蛋白胨肉汤培养基作为模拟物料,而在后续的干燥、分装工序采用流动性和密度都和头孢哌酮钠性质接近的PEG6000来作为模拟物料。
2.8.5 无菌工艺模拟验证实施过程
在无菌工艺模拟验证方案确定和培训之后,根据生产计划,选择生产间隙,安排了无菌工艺模拟验证,连续进行了3个批次模拟验证,批量比真实产品正常批量减少,为150kg。验证结束后,对相关设备进行了彻底清洁并灭菌。
2.8.6 样品培养
因为原料药工艺模拟验证后,需要培养的样品很多,一般的培养箱不能满足要求;因此,公司专门建设了具有温湿度控制功能的培养室。培养室在启用之前经过验证而且结果合格,在培养样品期间,一直有温湿度记录装置在线监控温湿度。
根据指南,选择分段培养条件:在20 ~ 25℃范围培养7天,然后在30 ~ 35℃范围培养7天。
样品培养后,在干燥工序和分装工序样品各发现一个阳性样品。
2.8.7 验证结果分析和评价
验证结果计算比较如下:头孢哌酮钠生产批量是350kg,在SFDA数据库查询用头孢哌酮钠生产的无菌制剂最大装量是3g,模拟验证中使用安慰剂150kg,所有的模拟验证样品检测后发现2CFU。
— 每批次污染水平就是2CFU,表明实际生产批次也是这个污染水平;
— 用这批原料药生产制剂最小单位数量计算:350000 g/3 g = 116667 units/batch;
— 计算CFU/unit最大值:2/116667 = 0.000017 CFU/unit。
结论:因为验证结果(0.000017 CFU/unit)比FDA指南推荐接受标准(0.0001 CFU/unit)低,因此,头孢哌酮钠无菌工艺模拟验证符合要求,通过验证。
2.8.8 验证过程中的偏差分析
经过详细调查和收集数据,在连续3批的无菌工艺模拟验证中,没有发生偏差情况,因此,没有涉及偏差处理问题。
尽管验证过程中没有发生操作偏差,但是最后样品培养结果显示有2个阳性样品,因此,公司按照MDD调查程序进行了调查,并针对调查后的原因进行了制度完善工作。
[1] FDA guidance for industry:sterile drug products produced by aseptic process-current GMP. September 2004.
[2] PIC validation of aseptic process. 2010.
[3] EU GMP annex 1:manufacture of sterile medicinal products,2008.
[4] PDA TR No.22 process simulation testing for aseptically filled products.
[5] PDA TR No.28 Process Simulation Testing for Sterile Bulk Pharmaceutical Chemicals.
[6] SFDA 《药品生产质量管理规范》草案(2010年修订).
猜你喜欢
现代畜牧科技(2021年8期)2021-10-13
山东冶金(2019年6期)2020-01-06
世界农药(2019年2期)2019-07-13
中国盐业(2018年20期)2019-01-14
现代园艺(2018年3期)2018-02-10
中华老年口腔医学杂志(2016年1期)2017-01-15
铜业工程(2015年4期)2015-12-29
中华皮肤科杂志(2014年4期)2014-12-19
中华皮肤科杂志(2014年3期)2014-12-19
中医研究(2014年5期)2014-03-11
