
溶胶-凝胶法制备TiO2/MCM-41负载型光催化剂及其性能表征
2011-01-29 08:03李杨薇宋卫坤
武汉科技大学学报 2011年5期
刘 红,石 飞,李杨薇,周 娟,宋卫坤
(1.武汉科技大学资源与环境工程学院,湖北武汉,430081;2.中国水利水电科学研究院水利研究所,北京,100048)
纳米TiO2制备方法大致有气相法和液相法,其中液相法包括沉淀法、水热合成法和溶胶-凝胶法等[1]。由于溶胶-凝胶法能够在常温条件下制备出纯度高、粒径分布均匀的材料,且实验过程易于控制[2],因此研究报道较多,但运用纳米TiO2粉体直接处理废水时,却存在吸附和沉淀效果差、回收再利用难等问题[3]。因此,制备比表面积大的负载型TiO2光催化剂,是提高TiO2光催化性能的有效途径。
自1992年Mobil公司首次合成介孔材料MCM-41以来[4],由于介孔材料具有比表面积大、孔隙率高和孔径分布窄等特点[5],因此在催化、吸附、分离等领域具有广阔的应用前景[6-7]。目前,利用分子筛比表面积大来提高光催化效果,将TiO2光催化剂引入介孔分子筛中成为该领域的研究热点。为此,本文以MCM-41分子筛为催化剂载体,采用溶胶-凝胶法制备TiO2/MCM-41负载型光催化剂,并通过XRD、BET、TEM、XPS对最佳条件下制备的光催化剂进行表征。
1 实验
1.1 原料
实验所用原料为钛酸丁酯、无水乙醇和冰乙酸,以上均为分析纯,分子筛为市售MCM-41。
1.2 实验方法
1.2.1 光催化剂的制备
将一定量的无水乙醇与7.5 m L钛酸丁酯在室温下进行混合,搅拌30 min后得到溶液A;将一定量的冰乙酸加入到8.75 m L无水乙醇中,充分混合后形成溶液B;将溶液B在磁力搅拌作用下缓慢滴加到溶液A中,混合均匀后得到溶液C。称取一定量的MCM-41粉体经80℃×4 h干燥后加入到溶液C中搅拌30 min,然后逐滴加入蒸馏水3 m L,搅拌直至胶凝。将所得的凝胶在80℃下烘干,并在一定温度下煅烧,冷却后将凝胶磨细,即得到TiO2/MCM-41光催化剂粉体。
1.2.2 光催化剂活性实验
将所制备的TiO2/MCM-41光催化剂以0.5 g/L的投加量投加到浓度为25 mg/L的甲基橙溶液中,超声分散5 min使其充分混合后,用高压汞灯进行0.5 h紫外光照反应。在反应进行的同时,通过磁力搅拌器高速旋转搅拌为反应提供溶解氧,使反应容器内气、固、液三相充分接触反应,随后分别取原水和经光催化反应的水样,离心分离后取上清液测定吸光度。
1.3 分析检测
吸光度采用UV-2550型紫外-可见分光光度计测定。根据Lambert-Beer定律,用降解前后测得的吸光度值来计算甲基橙的脱色率,计算式为
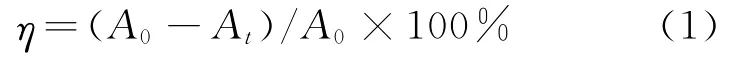
式中:η为脱色率,%;A0、At分别为初始吸光度和脱色t时刻后的吸光度。
XRD分析采用D/MAX-ШA型X射线衍射仪,Cu靶Kα射线,管压为40 k V,管流为40 m A;比表面积和孔隙分布分析采用SA3100型比表面积及孔隙分析仪;TEM和XPS分析采用H-9500型透射电子显微镜和PHI-5300型X射线光电子能谱仪。
2 结果与讨论
2.1 制备条件对TiO2/MCM-41活性的影响
调节不同的硅钛摩尔比,采用溶胶-凝胶法制备TiO2/MCM-41光催化剂,并用于降解初始p H值为6的甲基橙溶液,其结果如图1所示。由图1可看出,硅钛摩尔比对甲基橙脱色率的影响较大。随着Si含量的增加,脱色率逐渐增大,当硅钛摩尔比为1时,脱色率达到最大,之后脱色率随着Si含量的增加而减小。硅钛摩尔比较小时,催化剂的活性不高,主要是因为过量的TiO2分散在MCM-41的表面,堵塞其孔道,导致吸附量减少,从而影响光催化效果;过量的TiO2还会由于电子移位而不能负载在载体上,导致电子和空穴的复合,从而影响复合材料的光催化性能。而硅钛摩尔比大于1时,TiO2含量过低,电子和空穴的产量不足,也使得光催化活性下降。因此本研究确立适宜的硅钛摩尔比为1。

图1 硅钛摩尔比对TiO2/MCM-41催化剂活性的影响Fig.1 Effect of Si-Ti molar ratio on the photocatalytic activity of TiO2/MCM-41
将硅钛摩尔比为1的TiO2/MCM-41在不同温度下煅烧,然后进行光催化活性测试,其结果如图2所示。由图2可看出,样品活性随着煅烧温度的上升而增加,在700℃时样品活性达到最高,随后其活性逐渐降低。这是由于TiO2的主要晶型包括锐钛矿型和金红石型,金红石型的TiO2光生电子和光生空穴容易复合,使催化活性受到一定影响,但锐钛矿型光催化活性较高,当锐钛矿型和金红石型以适当比例共存时将会提高光催化活性[8]。随着煅烧温度的升高,锐钛矿型TiO2逐渐转变为金红石型,在700℃时体现出最佳的光催化活性,之后金红石型TiO2含量继续增大,反而使催化剂活性降低。因此本研究确定最佳煅烧温度为700℃。

图2 煅烧温度对TiO2/MCM-41催化剂活性的影响Fig.2 Effect of calcination temperature on the photocatytic activity of TiO2/MCM-41
图3为煅烧时间对TiO2/MCM-41催化剂活性的影响。由图3可看出,经过700℃×2 h煅烧后所制得的光催化剂活性最佳,甲基橙的脱色率达到93.7%。这是因为煅烧时间过短,TiO2晶化不完全,无法转化为具有较高活性的锐钛矿型;煅烧时间过长,会导致粉体发生团聚现象,材料的比表面积减小,导致催化活性下降。
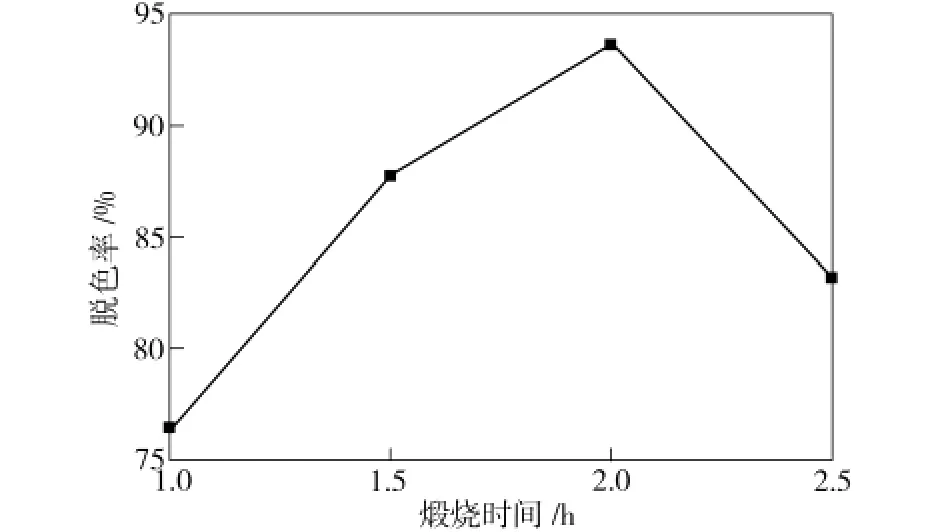
图3 煅烧时间对TiO2/MCM-41催化剂活性的影响Fig.3 Effect of calcination time on the photocatalytic activity of TiO2/MCM-41
2.2 TiO2/MCM-41表征
2.2.1 XRD分析
图4为MCM-41、TiO2/MCM-41和TiO2煅烧产物的XRD图谱。由图4可看出,TiO2/MCM-41和TiO2煅烧产物均出现了锐钛矿型特征衍射峰(对应的2θ为25.14°、37.88°、48.25°、62.72°)和金红石型特征衍射峰(对应的2θ为27.4°、36.1°、41.2°、54.3°),表明TiO2以及TiO2/MCM-41在700℃×2 h煅烧后均为金红石相与锐钛矿相的混晶结构。但由图4还可看出,TiO2中的晶相大部分为金红石型,TiO2/MCM-41中TiO2晶相大部分为锐钛矿型,这主要是因为MCM-41中的氧化硅可有效地抑制锐钛矿向金红石的转变[9-11]。因此,MCM-41不仅可以为TiO2的负载提供大比表面积,还可提高其晶相的转化温度和热稳定性。
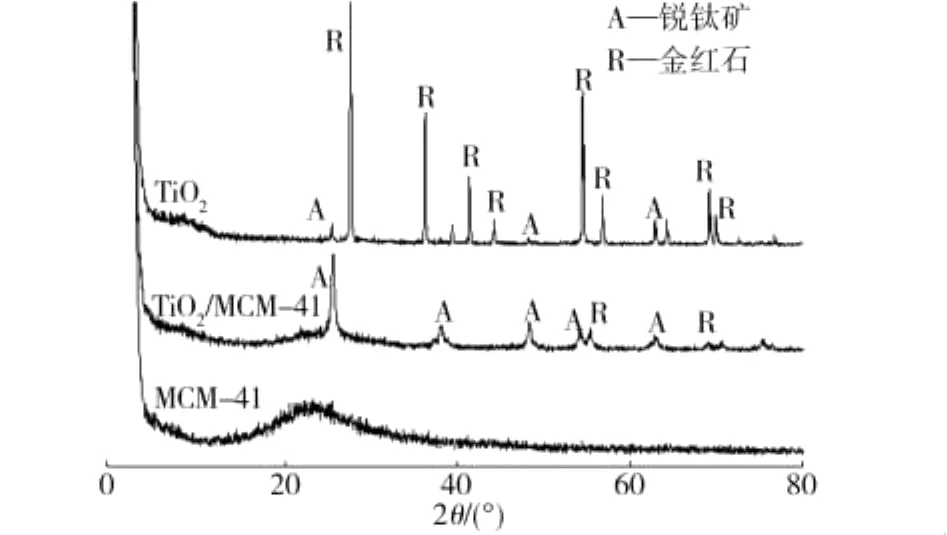
图4 MCM-41、TiO2/MCM-41和TiO2煅烧产物的XRD图谱Fig.4 XRD patterns of MCM-41,TiO2/MCM-41 and TiO2 calcined products
2.2.2 比表面与孔径结构
图5为TiO2/MCM-41的N2吸附-脱附曲线及孔径分布曲线。由图5可看出,样品的吸附-脱附等温线呈典型的IV型,且存在较大的滞后环,表明TiO2/MCM-41具有介孔结构[12]。通过多点BET法,利用相对压力为0.05~0.3所对应的氮吸附数据,计算出其粉体比表面积为358.8 m2/g。从氮脱附等温线计算得到孔径分布曲线看出,孔径主要集中在2~4 nm范围内,平均孔径为2.7 nm。通过BJH法,利用相对压力为0.975所对应的氮吸附数据,计算出总孔容积为0.429 m L/g。

图5 TiO2/MCM-41的N2吸附-脱附及孔径分布曲线Fig.5 Nitrogen adsorption-desorption isotherms and pore size distribution of TiO2/MCM-41
表1为MCM-41与TiO2/MCM-41的比表面积和孔结构数据。由表1可看出,负载后的TiO2/MCM-41比表面积有所下降,虽部分分子筛的孔径被堵塞,但样品仍保持较高的比表面积和孔容积,这样有利于将水中的污染物吸附至催化剂表面,同时有利于反应产物的迅速扩散,从而提高了光催化剂的活性。
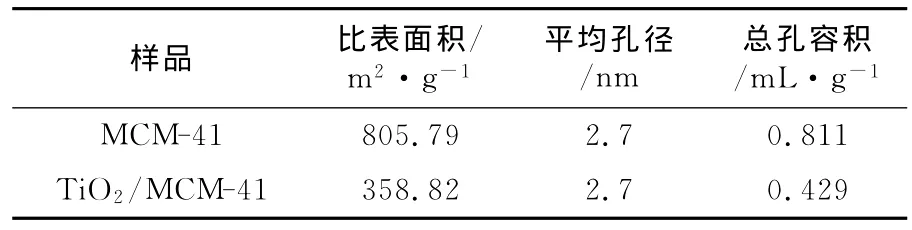
表1 样品的比表面积及孔结构数据Table 1 BET surface area and pore structure of MCM-41 and TiO2/MCM-41
2.2.3 TEM分析
图6为MCM-41与TiO2/MCM-41颗粒的TEM照片。由图6(a)可看出,MCM-41颗粒的表面有大量均匀规则的孔道。由图6(b)可看出,在MCM-41颗粒的表面负载了TiO2球形颗粒,产物晶粒粒径尺寸为20~25 nm。由于介孔材料MCM-41颗粒的孔径为2.7 nm,所以TiO2球形颗粒不能进入分子筛孔道均匀分布,大部分负载在分子筛表面,有些团聚在一起,这也是该样品比表面积小于分子筛的原因。

图6 MCM-41和TiO2/MCM-41颗粒的TEM照片Fig.6 TEM micrographs for MCM-41 and TiO2/MCM-41
2.2.4 XPS分析
图7为MCM-41与TiO2/MCM-41颗粒的XPS图谱。由图7可看出,在TiO2/MCM-41的XPS图谱中有明显的Ti2P峰。
图8为样品TiO2/MCM-41中Ti2P的XPS图谱,其电子结合能均用电子结合能为284.8 e V的ClS作为内标进行校正。由于电子的自旋轨道耦合,使Ti2P能级分解为Ti2P3/2与Ti2P1/2两个能级,其结合能分别为460.2、465.9 eV,与XPS标准数据手册中Ti4+的标准结合能[13](Ti2P3/2、Ti2P1/2分别为458.5、464.2 eV)基本一致,表明TiO2/MCM-41催化剂中的Ti是以+4价存在。

图7 MCM-41与TiO2/MCM-41颗粒的XPS图谱Fig.7 XPS spectra of MCM-41 and TiO2/MCM-41
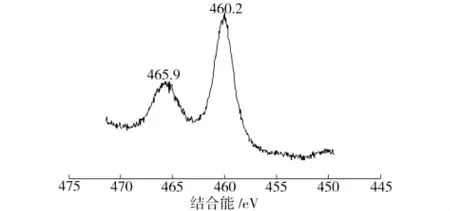
图8 TiO2/MCM-41中Ti2P的XPS谱图Fig.8 Ti2P XPS pattern of TiO2/MCM-41
图9为样品TiO2/MCM-41中O1S的XPS谱图。由图9可看出,经分峰拟合得到3个峰,结合能分别为531.4、533.2、534.5 e V。其中结合能较低的2个峰为TiO2的晶格氧,结合能较高的归为表面羟基氧[14],所占比率分别为62.0%、38.0%。由于表面羟基可以捕获光生电子,形成羟基自由基,所以该负载材料表现出良好的光催化性能。

图9 TiO2/MCM-41中O1S的XPS谱图Fig.9 O1S XPS pattern of TiO2/MCM-41
2.3 TiO2/MCM-41的光催化效果和回收利用
将所制光催化剂以0.5 g/L投加量加入到浓度为25 mg/L的甲基橙溶液中降解0.5 h,抽滤出光催化剂,用蒸馏水搅拌洗涤3次,干燥后进行二次利用实验。结果表明,TiO2/MCM-41光催化剂对甲基橙的首次利用和二次利用的脱色率分别为93.2%、30.4%。首次利用后光催化剂粉体的表面性质发生变化,光催化性能降低,但该光催化剂易自然沉降,回收率达到了76%,与自然沉降只有23%回收率的P25相比有较大的优势。因此TiO2/MCM-41光催化剂在催化完成后可用沉淀法从水中去除,这样可以避免光催化剂的流失和二次污染。
3 结论
(1)溶胶-凝胶法制备TiO2/MCM-41负载型光催化材料的最佳条件为:硅钛摩尔比为1,煅烧温度为700℃,煅烧时间为2 h。在此条件下制备的光催化剂降解甲基橙溶液0.5 h后,其脱色率为93.7%。
(2)TiO2/MCM-41中的氧化硅提高了锐钛矿相TiO2向金红石相转变的温度;TiO2/MCM-41的比表面积为358.8 m2/g,较MCM-41有所降低;MCM-41的表面负载了TiO2球形颗粒。
(3)TiO2/MCM-41催化剂中的Ti以+4价存在,TiO2表面羟基氧所占比率为38.0%。
[1] 谭怀琴,全学军,赵清华,等.TiO2光催化剂的制备与改性研究进展[J].材料导报,2005,19(专辑Ⅳ):59-61.
[2] 冀晓静.钛硅复合氧化物光催化剂的制备及光催化性能研究[D].东营:中国石油大学(华东),2008.
[3] 姜安玺,徐桂芹,相会强.光催化剂纳米TiO2的固定化技术研究进展[J].哈尔滨建筑大学学报,2002,35(4):43-46.
[4] Kresge C T,Leonowiez M E,Roth W J.Ordered mesoporous molecular sieves synthesized by a liquid crystal template mechanism[J].Nature,1992,359:710-712.
[5] 郑金玉,邱坤元,危岩.有机小分子模板法合成二氧化钛中孔材料[J].高等学校化学学报,2002,21(4):647.
[6] 刘红,粱晶,余薇,等.絮凝剂PTSS的分子结构研究[J].环境科学研究,2008,21(5):6-9.
[7] 冯利利,赵威,刘洋,等.MCM-41分子筛担载纳米TiO2复合材料光催化降解罗丹明B[J].物理化学学报,2009,25(7):1 347-1 351.
[8] 陈永,曹峰,马艳平.SiO2改性纳米TiO2及可见光催化性能研究[J].纳米科技,2008,5(3):40-43.
[9] Zhang Maolin,An Taicheng,Fu Jiamo,et al.Photocatalytic degradation of mixed gaseous carbonyl compounds at low level on adsorptive TiO2/SiO2photocatalyst using a fluidized bed reactor[J].Chemosphere,2006,64(3):423-431.
[10]冯春波,杜志平,赵永红,等.Au改性纳米TiO2材料对NPE-10光催化降解的活性[J].物理化学学报,2006,22(8):953-957.
[11]张青红,高濂,孙静.氧化硅对二氧化钛纳米晶相变和晶粒生长的抑制作用[J].无机材料学报,2002,17(3):415-421
[12]徐如人,庞文琴.分子筛与多孔材料化学[M].北京:科学出版社,2004.
[13]Wagner C D,Riggs W M,Davis L E,et al.Handbook of X-RAY photoelectro spectroscopy[M].New York:Perkin-Elmer Corporation,1979:453-460.
[14]Ho W K,Yu J C,Lee S C.Synthesis of hierarchical nanoporous F-doped TiO2spheres with visible light photocatalytic activity[J].Chem Commun,2006,10:1 115-1 117.
猜你喜欢
四川地质学报(2022年2期)2022-07-08
矿产勘查(2020年8期)2020-12-25
原子与分子物理学报(2020年5期)2020-03-17
——以金红石为例
中国金属通报(2020年23期)2020-03-15
济宁医学院学报(2018年6期)2018-12-28
无机盐工业(2017年6期)2017-03-11
湖南农业(2016年3期)2016-06-05
无机盐工业(2016年6期)2016-03-15
无机盐工业(2016年2期)2016-03-15
特产研究(2014年4期)2014-04-10
