
同位素内标-液相色谱-串联质谱法测定动物源性食品中氯霉素残留
2011-01-22 02:57:14陈君义孙慧宇舒永兰王云飞
化学分析计量 2011年2期
陈君义 孙慧宇 舒永兰 王云飞
(1.张家港出入境检验检疫局,张家港 215600; 2.徐州出入境检验检疫局,徐州 221006)
氯霉素(chloramphenicol,CAP)属广谱抗生素,曾作为动物临床常用的抗生素用于预防和治疗动物因细菌、立克氏体引起的炎症和疾病。但由于氯霉素具有抑制造血机能等毒副作用,还可引起不同的个体反应,如肝损伤、视神经炎、白血病等,因此目前许多国家严格禁止将氯霉素用于食用动物,规定其最高残留限量为0~0. 01 mg/kg。随着科学技术的进步,国外对进口动物源性产品中氯霉素的检出限量不断降低,欧盟的最低执行限量值为0.3 μg/kg[1]。有的国家甚至禁止对中国的某些动物源产品的进口,这对我国的出口贸易造成了严重影响,因此动物源性产品中氯霉素的残留检测变得尤为重要。
目前有关氯霉素在食品中的残留量分析方法主要有酶联免疫(ELISA)法[2]、气相色谱(GC)法[3]、气相色谱质谱联用(GC/MS)法[4]、液相色谱(HPLC)法[5]和液相色谱-串联质谱(LC-MS/MS)法[6-10]等。ELISA 法主要用于快速筛选检测,但定性和定量均不够准确;GC和GC/MS存在检出限难以达到要求、背景干扰较大或样品净化不完全以及衍生化难等问题;HPLC分析较准确且过程相对较简便,因而应用较多,但随着LC-MS/MS技术发展的日趋成熟,该方法被越来越多地应用于测定和确证动物源产品中氯霉素的痕量残留。以待测物的同位素标记物作为内标,补偿前处理过程和定量过程中的偏差,可以解决LC分析过程中的基质效应和处理过程中提取效率的问题[11]。
笔者在动物源性样品中加入氘代氯霉素作为内标,经乙酸乙酯提取、正己烷去脂后,无需过净化小柱,直接用LC-MS/MS测定动物源性产品中氯霉素残留量。该法操作简便,分析时间短,定性和定量的准确性高,满足进出口食品的兽药残留监控要求。
1 实验部分
1.1 主要仪器与试剂
液相色谱串联质谱仪(LC-MS/MS):API4000型,美国AB公司;
甲醇:HPLC级;
乙酸乙酯、正己烷:优级纯,国药集团;
氯霉素标准品(CAP):纯度为98.5%,德国Dr. E公司;
氯霉素-d5标准品(CAP-d5):纯度为98.6%,德国Witega公司;用色谱级甲醇配制成1 000 μg/L的标准储备液,贮存在4℃冰箱中,实验中根据需要用甲醇稀释至适当浓度的标准工作液。
实验用水为去离子水。
1.2 样品制备
在龙虾样品中取出有代表性的样品约500 g,取可食部分用粉碎机绞碎;从所取肠衣样品中取出有代表性样品约300 g,用剪刀剪碎(小于0.5 cm)。将粉碎的样品混合均匀,均分成两份,分别装入洁净容器作为待测样,密封,在-18℃条件下保存。
1.3 样品前处理
(1)龙虾:称取5 g样品(精确到0.01 g)置于50 mL具塞离心管中,准确加入200 μL氯霉素-d5内标溶液(10 ng/mL),加入20 mL乙酸乙酯,高速均质2 min,超声10 min,然后以3 500 r/min离心5 min,收集上清液,于45℃下旋转蒸发至近干,然后依次加入4 mL正己烷和5.0 mL水溶解,并转移至10 mL试管中。振荡1 min后离心,弃去有机相,取1 mL水相经0.45 μm滤膜过滤后上机测定。
(2)肠衣:称取5 g样品(精确到0.01 g)置于50 mL具塞离心管中,准确加入200 μL氯霉素-d5内标溶液(10 ng/mL),加入20 mL乙酸乙酯,于涡旋混合器上混匀1 min,超声波超声20 min,然后以3 500 r/min离心5 min,收集上清液,并按上述方法操作。
1.4 液相色谱与质谱条件
色谱柱:Agilent Eclipse XDB-C18柱(150 mm×4.6 mm,5 μm);柱温:25℃;流速:0.5 mL/min;进样量:25 μL;流动相:A相为甲醇,B相为水。
梯度洗脱程序:0~2 min,A相保持30%;2.0~3.5 min,A相从30%线性升至90%,保持3.5 min;0.1 min内,A相降至30%,保持至4 min。
电喷雾离子源,负离子扫描模式(ESI-),多反应监测(MRM);离子源喷雾电压:-4 500 V;离子源温度:550℃。
2 结果与讨论
2.1 样品前处理
目前在检测食品中氯霉素残留时通常采用乙酸乙酯提取,用正己烷去脂,然后再用乙酸乙酯反萃取等前处理方法。由于样品很难在均质器中达到完全破碎,尤其是肠衣,故在试验中,通过超声的方法改善提取效果,加入同位素内标,补偿提取偏差,免去小柱净化步骤,缩短了前处理时间,减少了有机试剂消耗量且有效保证了分析结果的稳定性和准确性。
2.2 质谱条件的优化
氯霉素分子式为C11H12O5N2Cl2,分子量为322,其同位素内标(5个不同位置上的氢被氘取代)的分子量为327,两者的理化性质接近,使用同位素可以矫正在样品提取过程中氯霉素的损失,提高测定方法的回收率。
将氯霉素标准品在负离子模式下进行质谱全扫描,其母离子为m/z320.8,主要子离子碎片有m/z151.8、 175.8、193.7、256.8(见图1)。根据丰度大小选择定性和定量离子对,见表1。
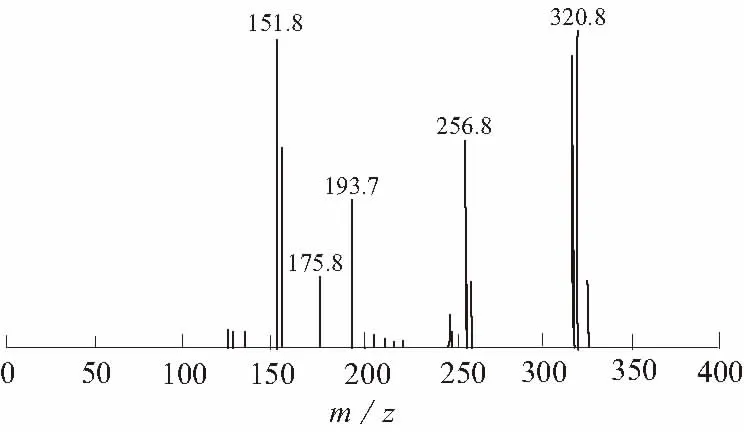
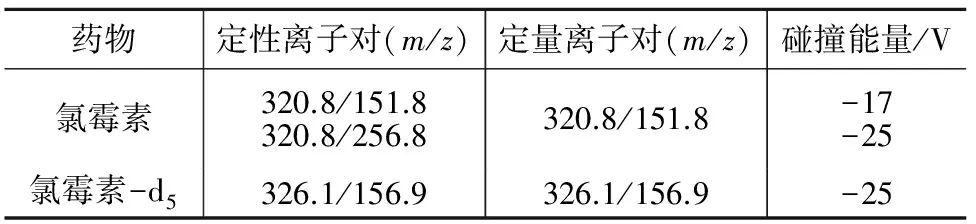
图1 氯霉素全扫描图
2.3 标准工作曲线
在标准系列溶液中加入40 μL(与样品中同位素内标体积比例相同)的同位素混合内标,按上述实验步骤操作,以峰面积比X(标准峰面积与内标峰面积之比)为横坐标,氯霉素浓度c(ng/mL)为纵坐标,绘制标准曲线,线性回归方程为c= 0.245 7X-0.001 3,r2=0.999 4,标准工作曲线线性范围为0.05~2.0 ng/mL。
2.4 样品中氯霉素含量的计算
样品处理后经液相色谱-串联质谱检测后,按照公式(1)计算样品中氯霉素的含量:
(1)
其中:X——样品中氯霉素的含量,μg/kg;
c——最终提取物中浓度,ng/mL;
V——最终定容体积,mL;
M——称样量,g。
按此方法检测,样品中氯霉素的检出限为0.05 μg/kg。
2.5 加标回收率和精密度试验
以不含有氯霉素残留的龙虾和猪肠衣为样品,按照1.3方法进行提取、净化和测定,在0.1、0.2、0.3 μg/kg 3个浓度水平上进行加标回收试验,氯霉素在两种样品中的添加回收率范围为71.8%~90.0%,相对标准偏差均小于3%,试验结果见表2。

表2 龙虾和肠衣样品中不同添加水平的回收率(n=5)
2.6 方法适用性
使用该方法对动物源性食品龙虾、银鱼、蚬肉、羊肠衣、猪肠衣、虾仁等进行检测,检测回收率稳定。
3 结论
采用氯霉素的同位素标记作为内标,补偿了处理过程和液质联用中产生的误差,有效地提高了定量的灵敏度,保证了实验的回收率;同时采用同位素内标,以双离子监控的模式,可以准确地甄别样品中氯霉素的存在,保证了检测结果的准确性,满足国内外对该类化合物在进出口食品中的监控要求。提取过程无需使用净化小柱,检测成本低,操作简便,分析时间短,实现了动物源性食品中对氯霉素的快速、经济检测。
[1] 2003/181/EC: Commission decision of 13 march 2003 amending decision 2003/657/EC as regards the setting of minimum required performance limits (MRPLs) for certain residues in food of animal origin[J].Official J, 2003,L71:17-18.
[2] 徐美奕,孟庆勇.养殖对虾中氯霉素残留的放射免疫分析[J].食品科学,2003,204(12):110-112.
[3] 宫向红,徐英江,张秀珍,等.水产品中氯霉素残留量气相色谱检测方法的探讨[J].食品科学,2006,27(7):222-224.
[4] 孙亮,陈海东.GC-MS/MS测定虾仁中氯霉素的研究[J].中国卫生检疫杂志,2003,13(3):293-294.
[5] 何方奕,李铁纯,李学程,等.固相萃取-高效液相色谱法测定肌肉中氯霉素的残留[J].食品科学,2006,27(12):629-630.
[6] 牛英娟,饶国瑛,李平,等.液相色谱/电喷雾离子阱质谱测定虾中氯霉素残留[J].分析化学,2004,32(12):1 698-1 698.
[7] Joe Storey, Al Pfenning, Sherri Turnipseed, et al. Determination of chloramphenicol residues in shrimp and crab tissues by electrospray triple quadrupole LC/MS/MS [J]. Laboratory Information Bulletin, 2003,19(6):412-431.
[8] Vytautas Tamosiunas, Julijonas Petraitis, Audrius Padarauskas. Chloramphenicol determination in milk by liquid chromatography-tandem mass spectrometry [J]. Chemija, 2006,17(2-3):25-29.
[9] 殷平,陈舜胜,邓晓军,等.液质联用检测水产品中氯霉素、氟苯尼考和甲砜氯霉素残留[J].现代食品科技,2007,23(10):83-87.
[10] 冯雷,尹丽珠,孙文通,等.禽畜肉中氯霉素残留量的液质联用分析方法[J].食品科学,2010,31(4):243-245.
[11] 黄挺,张伟,刘洋,等.液相色谱-同位素稀释质谱法测定配方奶粉中的烟酰胺[J].色谱,2007,25(6):922-925.
猜你喜欢
中国食品工业(2021年7期)2022-01-19 07:33:46
食品安全导刊(2021年20期)2021-08-30 06:40:34
中国动物检疫(2021年5期)2021-03-28 06:18:03
中国中医急症(2019年10期)2019-05-21 07:20:46
食品与机械(2017年4期)2017-07-05 14:46:35
家庭科学·新健康(2016年5期)2016-05-12 23:51:56
中国药物应用与监测(2015年5期)2015-12-11 03:15:53
川北医学院学报(2015年5期)2015-12-05 08:22:43
中国医疗美容(2015年1期)2015-07-12 10:06:55
中国药业(2014年21期)2014-05-26 08:56:34
