
高效液相色谱-质谱联用法测定人体血液中脂肪酸含量*
2011-01-22 01:31:06盛灵慧李保山
化学分析计量 2011年5期
许 倩 盛灵慧 李保山 黄 峥
(1.北京化工大学化工资源有效利用国家重点实验室,北京 100029; 2.中国计量科学研究院,北京 100013)
根据单双键组成脂肪酸可分为饱和脂肪酸、单不饱和脂肪酸和多不饱和脂肪酸3种。流行病学研究表明,脂肪酸与胰岛素抵抗、冠状动脉疾病、高血压、肥胖等慢性病以及乳腺癌、结肠癌等疾病的发生具有一定的关联性。人体血清中总脂肪酸的组成及其水平是人体摄入脂肪酸和代谢后的重要指标,进行人体血液中的脂肪酸组分分析有助于健康状况评价[1,2]。目前脂肪酸的测定通常采用衍生的方法对脂肪酸进行甲酯化反应后通过气相色谱或气相色谱-质谱法分析[3-5],但衍生化的过程不仅花费大量的时间,而且会对样品造成损失。因此笔者建立了一种快速、准确的定量方法,此方法使用Dole提取法[6],提取的样品直接进行分析,无需衍生化,用HPLC-MS分离、定量了人血液中游离脂肪酸,并采用氘代十六烷酸为内标物测定了回收率。
1 实验部分
1.1 主要仪器与试剂
液相色谱串联质谱联用系统(LC-MS-MS):包括高效液相色谱仪(Agilent 1200型,美国Agilent公司)和串联三重四极杆质谱仪(Agilent 6410型,美国Agilent公司);
电子分析天平:XP65型,d=0.01 mg,梅特勒-托利多仪器(上海)有限公司;
7种脂肪酸(十三烷酸、十四烷酸、十七烷酸、油酸、硬脂酸、二十烷酸和二十二烷酸)标准物质:中国计量科学研究院生物实验室制备;
11种脂肪酸(十二烷酸、十六烷酸、十六碳一稀酸、亚油酸 、十八碳三稀酸、花生酸、二十碳一稀酸、二十碳二稀酸、花生四稀酸、二十碳五稀酸、二十二碳六稀酸)标准物质:购自美国Sigma-Aldrich公司;
血清样品:解放军医学科学研究院;
甲醇:分析纯,美国J. T. Baker公司;
磷酸:分析纯,北京化学试剂公司;
实验所用其它试剂均为分析纯;
实验用水为millipore超纯水。
1.2 样品处理
取1 mL血清于15 mL离心管中,加入50 μL 同位素十六烷酸内标溶液(10 mg/L),加入5 mL混合液(异丙醇-正己烷-2 mol/L磷酸,体积比40∶10∶1),室温反应10 min,随后加入2 mL正己烷和3 mL水,涡旋混匀,以4 500 r/min离心5 min,提取全部上清溶液,氮气吹干。加入1 mL甲醇溶液复溶,进行HPLC-MS分析。
1.3 标准曲线配制
将18种脂肪酸分为饱和脂肪酸和不饱和脂肪酸两类,准确称量后配制了两组混合标溶液作为母液,溶液中每种脂肪酸浓度为200 μg/mL。从母液中吸取适量用甲醇稀释成0.2~100 μg/mL的标准工作液,存放在4℃冰箱中备用。
1.4 仪器工作条件
(1)色谱条件
色谱柱:XTerra C8(2.1 mm×150 mm,3.5 μm),美国Waters公司;流动相A为水(含5 mmol/L乙酸铵),B为乙腈(含5 mmol/L乙酸铵),流速为0.1 mL/min,流动相比例见表1。
(2)质谱条件
使用电喷雾电离源(ESI)负离子模式和多反应监测(MRM)模式分析;喷雾气(Gasl)、雾化辅助加热气(Gas2)、气帘气(CUR)和碰撞气(CAD)均为氮气;裂解电压:130 V;干燥气温度:350℃;脱溶剂气流速:9.0 L/min;毛细管电压:3.8 kV。
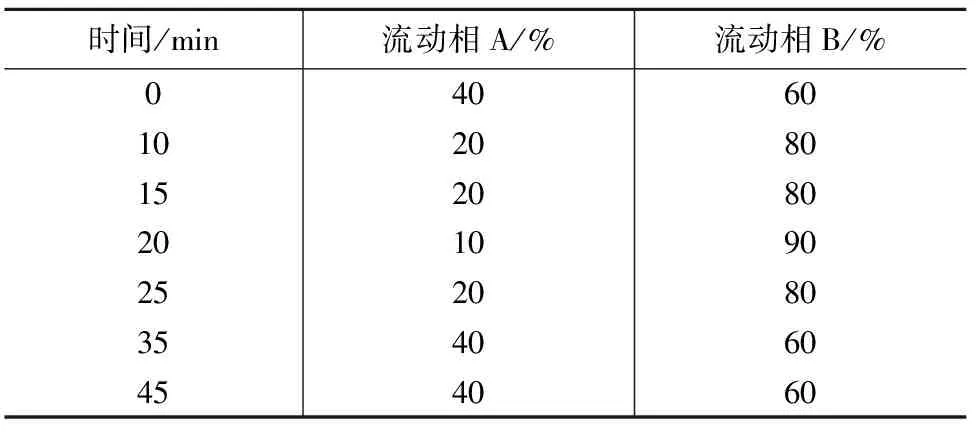
表1 流动相配比
2 结果与讨论
2.1 仪器条件的建立
脂肪酸分析用的MS-MS多反应监测(MRM)模式应尽量减少血浆基质干扰。脂肪酸在负离子模式下,加碰撞能产生的碎片离子灵敏度很低。最大碎片离子为脂肪酸失去羟基水得到的 [M-18]-离子,但这个碎片离子的灵敏度很低,无法满足定量条件的要求。虽然选择离子扫描(SIM)模式比MRM模式在脂肪酸的分析中有更好的灵敏度,但是SIM模式会有更多的分析干扰。为了减少血浆基质的干扰,同时确保有较好的灵敏度,在分析时仍采用MRM模式,但不加碰撞能,子离子依然选择母离子碎片离子。
2.2 流动相的选择优化
考察了含有不同比例乙腈-水的多种流动相体系。在纯水和乙腈为流动相时,脂肪酸离子化效率较差,质谱响应很弱。在流动相中添加5 mmol/L乙酸铵后,改变了流动相的pH值,提高了离子化效率,响应值增加。实验以5 mmol/L乙酸铵乙腈-水溶液为流动相,采用梯度洗脱程序,实现了18种脂肪酸良好分离,且峰形良好。
2.3 提取条件优化
试验考察了提取混合液加入量对样品中脂肪酸提取效果的影响。分别取1 mL血清于15 mL离心管中,加入50 μL同位素十六烷酸内标溶液(10 mg/L),分别加入1、3、5、7、9 mL混合液(异丙醇-正己烷-2 mol/L磷酸,体积比40∶10∶1),在室温下反应10 min,加入2 mL正己烷和3 mL水,涡旋混匀,以4 500 r/min离心5 min,提取全部上清溶液,氮气吹干。加入1 mL甲醇溶液复溶,进行HPLC-MS分析。
试验发现,随着混合液加入量的增加,样品提取的回收率也增加,但当提取液到5 mL后,样品回收率增加很少并趋于恒定,所以最终选取加入5 mL混
合液提取样品。
2.4 色谱柱的选择
试验比较了XTerra C8色谱柱(2.1 mm×150 mm,3.5 μm ),XTerra C18色谱柱(2.1 mm×150 mm,3.5 μm )和BEH C18色谱柱(2.1 mm×50 mm,1.7 μm)在相同的梯度条件下,对标准溶液18种脂肪酸的分离效果。结果表明,XTerra C8色谱柱对18种脂肪酸的分离效果最好,见图1。因此选用XTerra C8色谱柱进行样品分析。BEH C18色谱柱是UPLC分离中的通用型色谱柱,但对于脂肪酸这种非水溶性的弱极性化合物,分离度较差;XTerra C8色谱柱比XTerra C18色谱柱极性弱,因此对脂肪酸这种弱极性化合物有更好的分离效果。
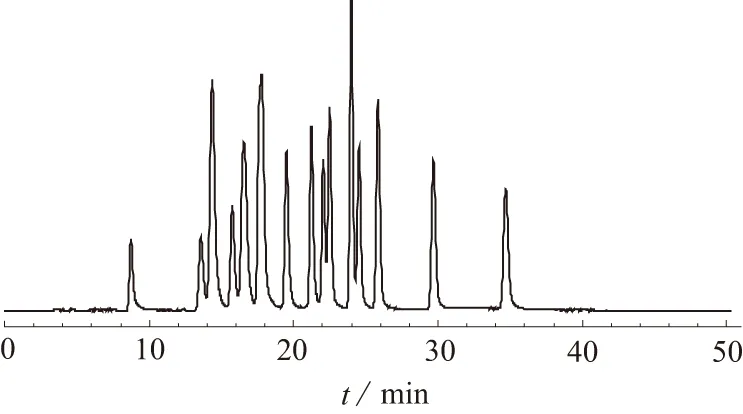
图1 XTerra C8色谱柱液质谱图
2.5 线性方程及检出限
在0.2~200 μg/mL浓度范围内,以外标物峰面积y对浓度x(μg/mL)绘制标准曲线,并按S/N=3计算检出限,S/N=10计算定量限,结果见表1。由表1可见,18种脂肪酸在测定范围内均呈良好的线性关系,相关系数均在0.99以上。

表1 18种脂肪酸线性方程及检出限
2.6 方法回收率及精密度
在1 mL血清中(本底值均为0)加入50 μL同位素十六烷酸内标溶液(10 mg/L),用本法提取并测定,以考察方法的回收率。试验表明,方法的回收率为90.02%~100.05%,结果见表2。
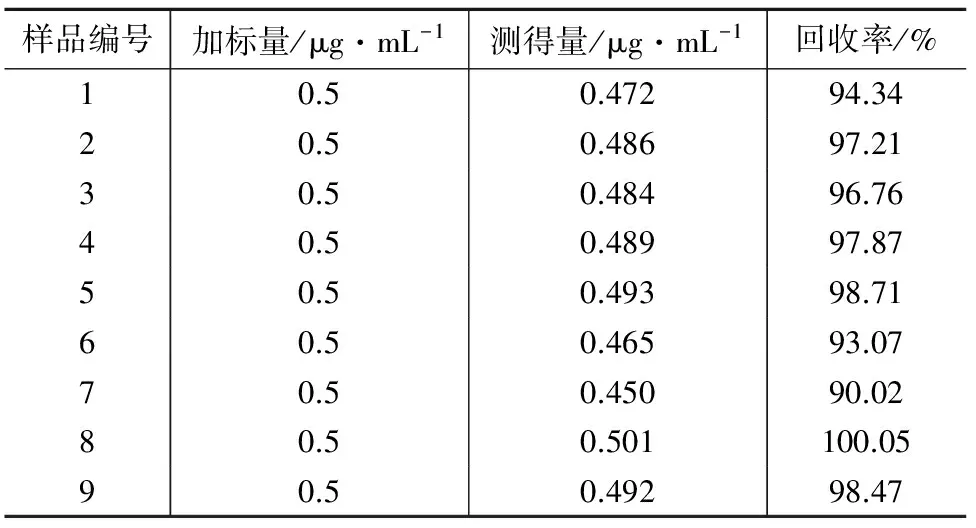
表2 同位素十六烷酸加标回收试验结果
为考察方法的精密度,分别平行提取9个血清样品,每个血清样品重复测定3次,结果列于表3。

表3 血清中脂肪酸含量及实验精密度
由表3可见,方法精密度在9%以内,因此该法可以用于血清样品中游离脂肪酸的测定。
3 结语
建立了一种高效液相色谱-质谱联用法测定人体血液中脂肪酸含量的方法。该法快速、准确,具有精密度高、准确度好、操作简单等特点,为人类相关疾病的脂肪酸分析提供了一种可靠、高效的方法。
[1] Michael Puttmann, Harald Krug, Elke von Ochsenstein, et al. Fast HPLC determination of serum free fatty acids in the picomole range[J]. Clinical Chemistry,1993,39(5):825-832.
[2] Sebastien Gagne, Sheldon Crane, Zheng Huang, et al. Rapid measurement of deuterium-labeled long-chain fatty acids in plasma by HPLC-ESI-MS[J]. Journal of Lipid Research,2007,48:252-259.
[3] Seppanen Laakso T, Laakso I, Hiltunen L R. Analysis of fatty acids by gas chromatography and its relevance to research on health and nutrition[J]. Anal Chim Acta, 2002,465:39-62.
[4] Cyrous O Kangania,David E Kelleyb, James P DeLanya. New method for GC/FID and GC-C-IRMS analysis of plasma free fatty acid concentration and isotopic enrichment[J]. Journal of Chromatography B,2008,873:95-101.
[5] Hyun Jin Jung,Won Yong Lee,Bong Chul Chung. Mass spectrometric profiling of saturated fatty acid esters of steroids separated by high-temperature gas chromatography[J]. Journal of Chromatography A, 2009,1 216:1 463-1 468.
[6] Michael Puttmann, Harald Krug, Elke von Ochsenstein, et al. Fast HPLC Determination of Serum Free Fatty Acids in the Picomole Range[J]. Clinical Chemistry,1993,39(5):825-832.
[7] 李海静,吴胜明,方均建,等. 气质联用法测定人血清游离脂肪酸[J]. 质谱学报,2009,30(2):83-87.
猜你喜欢
食品安全导刊(2021年20期)2021-08-30 06:39:48
中国生殖健康(2020年5期)2021-01-18 03:00:06
中国生殖健康(2018年5期)2018-11-06 07:15:56
当代化工研究(2016年5期)2016-03-20 16:21:35
中国塑料(2015年6期)2015-11-13 03:03:11
中国医药科学(2015年5期)2015-08-01 14:14:51
中国洗涤用品工业(2015年8期)2015-02-28 19:02:49
特产研究(2014年4期)2014-04-10 12:54:22
Sciences in Cold and Arid Regions(2014年6期)2014-03-31 00:28:31
色谱(2013年6期)2013-07-13 05:24:12
