
干冷空气淬火制备钒酸锂材料性能研究*
2011-01-22 02:17:47,,
无机盐工业 2011年9期
,,
(西南林业大学西南山地森林资源保育与利用重点实验室,云南昆明 650224)
LiV3O8作为锂二次电池潜在的正极材料,最早的合成方法是固相法,即将物质的量比为1∶3的Li2CO3和V2O5混合研磨后在680 ℃下直接固相烧结[1]。这种方法合成的LiV3O8结晶度高,锂离子扩散路径长,极化电阻大,实际放电容量约为180 mA·h/g,远低于理论值287 mA·h/g。此外,该方法因煅烧温度高于物料的熔点,部分原子挥发,产物中锂、钒确切原子比难控制,而且熔融产物会污染反应容器。液相反应可以实现在低温下合成LiV3O8,如水热法[2]、溶胶-凝胶法[3]等。这类方法合成的钒酸锂中锂、钒原子比例容易控制,产物的结晶度较低,比表面积较大,具有较高的比容量,但可能因为没有形成完整的层状结构,循环稳定性不理想。此外,液相合成法步骤繁琐,反应时间长,不适合大规模生产。一些后处理方法[4-5]被用来克服直接固相合成法的缺点,如超声波处理、有效球磨、在层间插入无机小分子等。这些后处理方法通常需要经过过滤、洗涤、烘干、热处理等步骤,增加了制备工序。A.Yu等[6]将650 ℃固相熔融产物迅速置于冷水中淬火,在不同温度热处理得到结晶度不同的钒酸锂样品。这种方法仍然不能克服熔融产物对反应容器的污染,且冷却后的熔融物坚硬难研磨。笔者以Li2CO3和V2O5作反应物,在600 ℃下固相烧结得到半熔融物,将此半熔融物迅速置于干冷空气中淬火获得LiV3O8样品。这种方法简单易行,省时节能,不会对反应容器造成污染,产物呈疏松多孔状,容易处理,其电化学性能也较好。
1 实验
1.1 样品的制备
将物质的量比为1∶3的Li2CO3和V2O5在玛瑙研钵中充分研磨混匀,在压片机上压制成厚约0.2 cm的圆片。将此圆片置于SCQ-8-12B型管式气氛炉的干燥气氛中,加热至600 ℃烧结,升温速度为1 ℃/min。当炉温升至600 ℃时,将半熔融物取出并迅速置于事先准备好的干冷空气中淬火。干冷空气是将一个密封良好的不锈钢容器放在冰水混合物中得到的。将淬火后的产物在玛瑙研钵中研细,于120 ℃真空干燥4 h得到样品a。作为参照样,样品b和c是反应物在气氛炉中于550 ℃和680 ℃下分别煅烧8 h,然后随炉自然冷却至室温后得到。
1.2 测试与表征
采用荷兰Philips公司X′pert Pro MPD型X射线衍射仪(Cu靶Kα射线,λ=0.154 056 nm,扫描范围2θ=10~70°)分析样品的结构;用荷兰Philips-FEI公司XL30 ESEM-TMP环境扫描电子显微镜(ESEM)分析样品的形貌;用武汉金诺电子有限公司LAND CT2001 A型电池测试系统对电池进行容量及循环性能测试。
1.3 电池的制备及性能测试
将制得的活性材料与超导炭黑、黏结剂聚四氟乙烯(PVDF)按85∶10∶5的质量比称量,再加入N-甲基吡咯烷酮(NMP),搅拌均匀后涂在铝集流体上干燥制成正极,以金属锂片为负极,采用Celgard 2320型隔膜,电解液是1 mol/L的LiPF6/EC-DMC(1∶1),制备成CR2025纽扣电池。充放电测试采用LAND电池程控测试仪(LAND_060331型),电压范围为2.0~4.0 V。循环伏安实验在CHI660C电化学工作站上进行,扫描速度为0.1 mV/s。电池组装在充满氩气的手套箱中完成,测试在室温下进行。
2 结果与讨论
2.1 LiV3O8的结构和形貌



图1 不同条件下合成的LiV3O8样品的XRD谱图表1 不同条件下合成的LiV3O8样品的晶胞参数和I(100)/I(003)及I(100)/I(020)值
图2是不同条件下合成的LiV3O8样品的SEM照片。由图2可知,样品a的颗粒大小和形貌与样品b和c明显不同。样品a的形貌为均匀的棒状颗粒,宽为0.3~1 μm,长为1~2 μm。样品b由大的块状颗粒组成,其中有些颗粒畸变。样品c由大的薄片组成,具有明显的各向异性。不同样品形貌和粒径的差别可能与界面和孔隙间的有质动力有关[8],这种有质动力能促使晶体快速生长,消除颗粒间的孔隙。在层状LiV3O8的形成过程中,有质动力的影响使VOn多面体沿(100)晶面方向快速生长,通过淬火骤冷处理,样品a中晶体沿(100)方向的生长被中止,因而不同于样品b和c的较大颗粒和片状结构,样品a颗粒较小。

图2不同条件下合成的LiV3O8样品的SEM照片
2.2 LiV3O8的电化学性能
图3给出了不同样品在0.25 C倍率下的循环性能。从图3可以看出,3种样品经过30次循环后容量都有不同程度的下降,其中样品a在整个循环中容量最高,它的第2、10、30次放电容量分别为256、227、189 mA·h/g,表现出较好的循环稳定性。需注意的是,样品b的循环曲线呈弯弓状,即在开始的几个循环中它的放电容量逐渐增大,但在约15次循环后容量迅速衰减。这种现象可能与样品b的结构和形貌有关。样品b颗粒比较致密,颗粒间的孔隙少,有效比表面积较小,不利于电解质溶液与活性物质间的充分浸泽,锂离子的扩散受阻,因而初始容量较低。随着循环的进行,电解液和活性物质间的接触得到改善,容量逐渐升高。但因为样品b中有杂相存在,且层状结构不够完整,因此循环稳定性不好。样品c是按照传统方法合成的,它的循环稳定性与样品a相似,但每次循环平均放电容量比样品a低约40 mA·h/g,即放电容量只有样品a的80 %左右,这种现象与它的颗粒大、比表面积小有关。

图3不同条件下合成的LiV3O8在0.25 C充放电倍率下的循环性能
图4A比较了不同样品在0.25 C倍率下第10次循环的充放电曲线。从图4A看出,样品a的容量最高,为227 mA·h/g,样品c的容量最低,为188 mA·h/g。这是因为,与样品c相比,样品a的结晶度低,有效比表面积大,有更多的嵌脱锂位置。图4B是样品a和c的dQ/dV曲线。由图4B看出,样品a具有同样品c相似的dQ/dV曲线图,它们的主要差别在2.0~2.9 V电压区间。在此电压范围,两种样品都有4对充放电峰,总的来说,样品a的充放电峰面积大于样品c,意味着样品a有更高的充放电容量。样品a位于约2.3 V处的放电峰比样品c的明显,表明样品a在放电过程中有Li5V3O8形成。
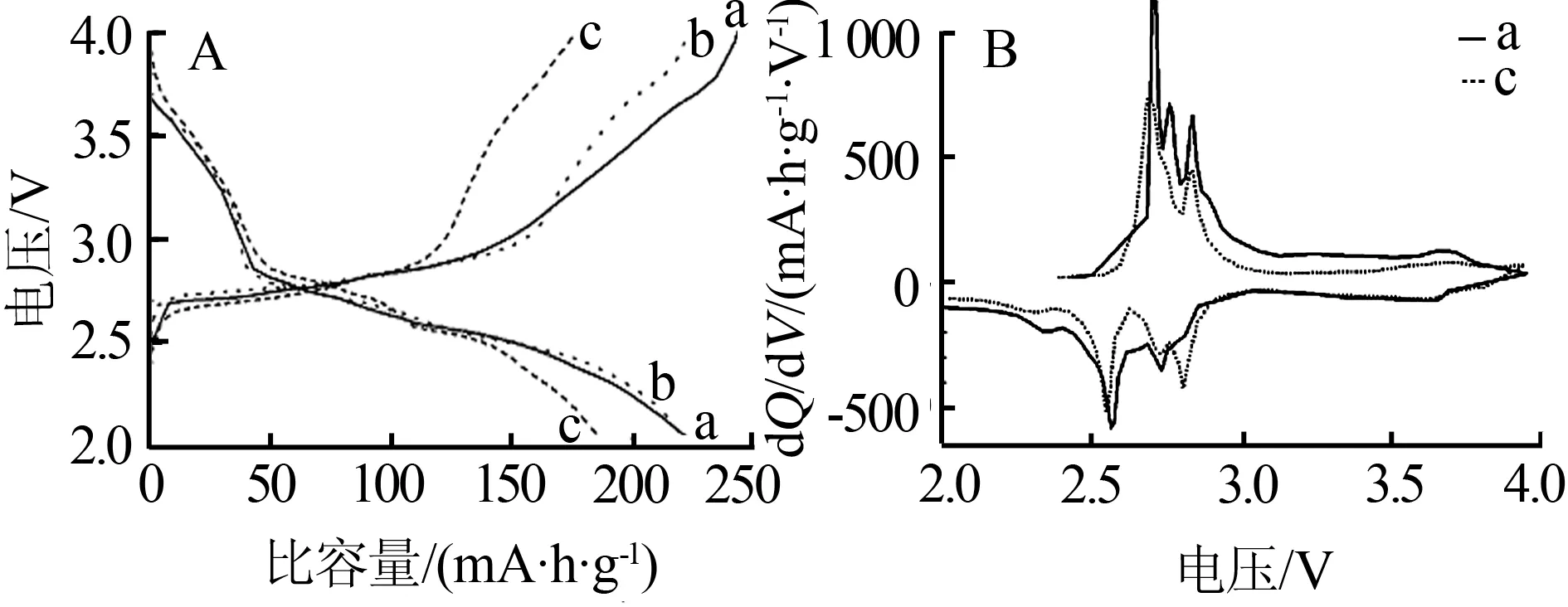
图4不同样品在0.25 C充放电倍率下第10次循环的(A)充放电和(B)dQ/dV曲线图
图5A比较了样品a和c第2次循环伏安曲线。由图5A看出,样品c位于2.85 V处出现一个主要氧化峰,两个主要还原峰分别位于2.76 V和2.48 V处。在此电压范围内,样品a有多个分裂的还原峰,表示锂离子嵌入样品a的不同位能处。循环伏安曲线上出现的所有峰都源自于锂离子嵌脱时Li1+xV3O8中不同x值(0 图5样品a和c第2次循环伏安曲线(A)和样品a第2次和第7次循环伏安曲线(B) (A和B扫描速度分别为0.1 mV/s和1 mV/s) 图6是样品a和c两次循环后截止电压为2.8 V时的交流阻抗图(EIS),图中内嵌的是EIS谱的等效电路图[9],表2是采用Zsimpwin软件拟合处理得到的电化学元件参数。等效电路中Rb为溶液电阻和接触电阻;Rf和Cf分别为覆盖在电极表面的固体电解质膜层(SEI)的阻抗和容抗;Q为与双电层电容相关的恒相位角元件,用于描述多孔电极产生的压缩半圆;Rt为电极反应的电荷转移电阻;ZW为锂离子的半无限Warburg扩散阻抗;Cint为锂离子在晶格中累积产生的电容。从图6可知,两种样品EIS图形状相似,都由中高频区的两个半圆和一条低频区的直线组成。其中第一个半圆反映了电极表面上的膜电阻Rf和膜电容Cf;第二个半圆反映了锂离子在电极材料和电解液界面处的迁移过程,由电荷转移电阻和双电层电容组成,用Rt和Qt表示,相应的nt表示Qt中电容成分的大小,当nt数值接近于1时,相应电化学元件接近于纯电容元件的特征;直线则代表Warburg阻抗。从表2可知,样品a的Rb、Rf、Rt和ZW都比样品c的小,尤其Rt值小约40 %,表明样品a的接触电阻、膜电阻和电荷转移电阻都较小,因此锂离子在样品a中的嵌脱反应更容易。 图6 样品a和c经2次循环后截止电压为2.8 V时EIS图表2 相应于图6的等效电路拟合参数值 以Li2CO3和V2O5为原料,在600 ℃下固相烧结,将得到的半熔融物在干冷空气中淬火处理合成了纯相的LiV3O8样品。通过XRD和SEM分析得出,淬火骤冷处理能抑制晶体继续生长,获得结晶度较低、具有一定程度各向同性的产物,这种产物颗粒较小,且颗粒间有较多的孔隙。电化学循环测试结果表明,与直接固相合成样品相比,这种方法合成的样品比容量和循环性能都有提高。EIS结果表明,淬火处理样品电化学性能较好的主要原因是此样品中锂离子在电解液/电极界面的传质电阻较小。 [1] Wadsley A D.Crystal chemistry of non-stoichiometric pentavalent vanadium oxides:crystal structure of Li1+xV3O8[J].Acta Crystallographica,1957,10:261-267. [2] Xu H,Wang H,Song Z,et al.Novel chemical method for synthesis of LiV3O8nanords as cathode materials for lithium ion batteries[J].Electrochim.Acta,2004,49(2):349-353. [3] Dubarry M,Gaubicher J,Moreau P,et al.Formation of Li1+nV3O8/ β-Li1/3V2O5/C nanocomposites by carboreduction and the resulting improvement in Li capacity retention[J].J.Electrochem.Soc.,2006,153(2):A295-A300. [4] Kumagai N,Yu A.Ultrasonically treated LiV3O8as a cathode material for secondary lithium batteries[J].J.Electrochem.Soc.,1997,144(3):830-835. [5] Manev V,Momchilov A,Nassalevska A.A new approach to the improvement of Li1+xV3O8performance in rechargeable lithium batteries[J].J.Power Sources,1995,54(2):501-507. [6] Yu A,Kumagai N,Liu Z,et al.A new method for preparing lithiated vanadium oxides and their electrochemical performance in secondary lithium batteries[J].J.Power Sources,1998,74(1):117-121. [7] Kawakita J,Miura T,Kishi T.Lithium insertion and extraction kinetics of Li1+xV3O8[J].J.Power Sources,1999,83(1/2):79-83. [8] Panneerselvam M,Rao K J.Novel microwave method for the synthesis and sintering of mullite from kaolinite[J].Chem.Mater.,2003,15(11):2247-2252. [9] 吕东升,李伟善.尖晶石锂锰氧化物锂离子嵌脱过程的交流阻抗谱研究[J].化学学报,2003,61(2):225-229.


3 结论
猜你喜欢
少年博览·小学高年级(2023年3期)2023-05-30 10:48:04
中学生数理化·中考版(2022年12期)2022-02-16 07:36:52
制造技术与机床(2017年3期)2017-06-23 08:11:52
制造技术与机床(2017年3期)2017-06-23 08:11:50
电子制作(2016年23期)2016-05-17 03:53:41
肇庆学院学报(2016年5期)2016-03-11 18:09:20
中国塑料(2015年7期)2015-10-14 01:02:40
新疆钢铁(2015年3期)2015-02-20 14:13:56
汽车与新动力(2014年5期)2014-02-27 12:10:48
化学分析计量(2013年5期)2013-03-11 16:37:51
