
萘在贵金属Pd、Pt及Pd-Pt催化剂上的加氢活性及耐硫性能
2010-11-06 07:01张小菲邵正锋毛国强何德民张秋民梁长海
物理化学学报 2010年10期
张小菲 邵正锋 毛国强 何德民 张秋民 梁长海
(大连理工大学化工学院精细化工国家重点实验室,辽宁大连 116012)
萘在贵金属Pd、Pt及Pd-Pt催化剂上的加氢活性及耐硫性能
张小菲 邵正锋 毛国强 何德民 张秋民*梁长海*
(大连理工大学化工学院精细化工国家重点实验室,辽宁大连 116012)
采用等体积浸渍法制备了SiO2-Al2O3负载的Pd、Pt单金属催化剂及Pd/Pt摩尔比分别为1∶1、1∶4、4∶1的双金属催化剂(Pd1Pt1、Pd1Pt4、Pd4Pt1),对其进行X射线衍射(XRD)、透射电镜(TEM)、CO化学吸附和X射线光电子能谱(XPS)表征,并详细考察了各催化剂的萘加氢活性和耐硫性能.结果表明,在实验考察范围内,Pd4Pt1催化剂上的萘转化率最高可达98.2%,全饱和产物十氢萘选择性最高可达93.6%,十氢萘反/顺生成率之比最高可达7.8,均高于单金属Pd(97.5%,59.1%,4.3)和Pt(96.8%,39.9%,2.9)催化剂的值.萘在三种催化剂上的加氢速率顺序为vPd4Pt1>vPd>vPt.添加二苯并噻吩(DBT)后Pd4Pt1上的萘转化率和十氢萘选择性仍然最高,十氢萘反/顺比在Pt催化剂上不受影响,在Pd4Pt1催化剂上稍有降低,而在Pd催化剂上降低明显.在三种不同Pd/Pt摩尔比的双金属催化剂中,Pd4Pt1催化剂上的萘转化率和十氢萘选择性在添加DBT前后都是最佳的.
贵金属; 萘; 加氢; 耐硫性; 十氢萘反/顺生成率之比
随着石油的重质化和劣质化趋势日益严重及环保法规的日趋严格[1-2],降低油品中的芳烃含量显示出极其重要的现实意义.油品中过多的芳烃不但会降低柴油的十六烷值,增高喷气燃料的烟点,也会增加油品燃烧过程中污染气体及颗粒物的排放.加氢脱芳(HDA)是目前工业上脱除油品芳烃的最主要手段.因此研究开发高活性的加氢脱芳催化剂一直是研究的热点.工业上加氢脱芳通常采用深度脱硫——芳烃饱和两段工艺,在第一段采用金属硫化物催化剂,通过苛刻加氢处理,将硫含量降低,在第二段用贵金属催化剂进行芳烃饱和.但贵金属对原料中的硫化物非常敏感,极易中毒失活.因此提高贵金属催化剂的耐硫性能对工业生产将有重要意义.Pd-Pt双金属催化剂以其改善耐硫性的突出作用成为长期以来的研究热点.将Pd引入负载型Pt催化剂有利于催化剂抗硫性提高的文献早有报道[3-6]. Yasuda和Yoshimura[3]证实在USY分子筛中Pd-Pt合金的形成提高了四氢萘的加氢活性和催化剂抗硫性,当Pd/Pt摩尔比为4∶1时,抗硫性达到最大值.他们将Pd、Pt系统的高抗硫性归因于电子效应和结构效应.Koussathana等[4]也得出了相同的结果. Fujikawa等[5]对 Pt-Pd/SiO2-Al2O3催化剂进行了研究,提出当Pd与Pd+Pt的质量比为0.7时催化剂的抗硫性达到极大值.Lin等[6]研究认为除了电子效应外Pd的添加还能够有效抑制Pt粒子的团聚,从而提高了抗硫性能.
萘在贵金属催化剂上的加氢产物主要包括四氢萘和反-、顺-十氢萘.反-十氢萘的热稳定性优于长链烷烃,是提高航空煤油热稳定性必不可少的添加组分;顺-十氢萘主要用于生产癸二酸进而生产尼龙6、尼龙10和增塑剂[7].顺-十氢萘受热力学平衡的影响[8],或者通过在贵金属催化剂活性位上加、脱氢立体异构还可进一步生成反-十氢萘[9].产物中十氢萘的反/顺比与催化剂的种类、担载量、合成方式等有很大的关系[10-14],例如Rh作萘氢化催化剂时主要生成顺-十氢萘[10-12],Pt作催化剂时主要生成顺-十氢萘,而Pd作催化剂时主要是生成反-十氢萘[8,13-14].因此,反应最终产物(顺式和反式十氢萘)的组成也可作为催化剂氢化能力的测量依据.本实验采用SiO2-Al2O3担载的Pd、Pt及Pd-Pt双金属催化剂,以萘为模型化合物,比较了各种催化剂的芳烃加氢性能和耐硫性,考察了不同Pd/Pt摩尔比对催化剂活性和耐硫性的影响及不同催化剂对产物十氢萘顺反组成的影响.
1 实验部分
1.1 催化剂的制备
将工业级SiO2-Al2O3(Al与Si原子比为4.8)载体于空气中500℃焙烧3 h,所得载体BET比表面积为247 m2·g-1,孔容为0.62 cm3·g-1.
Pd、Pt(担载量0.5%,质量分数,下同)单金属催化剂采用等体积浸渍法制备,具体过程如下:分别将一定量PdCl2(AR)或PtCl4(AR)溶于1 mol·L-1的盐酸(AR)溶液中配成浸渍溶液,加入适量SiO2-Al2O3载体,再将样品移入蒸发皿在烘箱中30℃放置3 h,110℃空气中干燥3 h,300℃氧气和氩气混合气(V(O2)/V(Ar)=5)中焙烧4 h,最后于300℃氢气中还原4 h.Pd-Pt双金属催化剂采用等体积共浸渍法制备:保持总担载量0.5%不变,分别按Pd/Pt摩尔比为4∶1、1∶1、1∶4,将PdCl2和PtCl4溶于1.0 mol·L-1的盐酸溶液中配成浸渍溶液,加入SiO2-Al2O3载体,后续浸渍与制备方法同前.
1.2 催化剂的表征
采用日本Rigaku公司生产的D/MAX-2400型X-射线衍射仪测定(Cu Kα辐射源,Ni滤波,管电压40 kV,电流100 mA)催化剂的晶相.采用透射电镜(JEM-100CXII,日本JEOL公司)观察催化剂的形貌,加速电压为180 kV;测试前,样品在乙醇中用超声波处理5 min,然后滴在铜网上,待乙醇挥发后分析样品.采用美国康塔(QuantaChrome)公司产的CHEMBET 3000化学吸附仪测定催化剂的化学吸附量;先将催化剂在300℃下用H2还原2 h,再在He气氛下吹扫2 h,并降温到30℃;再注入CO气体直至饱和吸附,得到样品化学吸附量.使用Thermo ESCALAB 250型X光电子能谱仪(美国Thermo Fisher Scientific公司)进行X射线光电子能谱(XPS)分析,采用单色化的Al Kα(Mono Al Kα)源辐射作为X射线激发光源,其参数为:1486.6 eV,150 W,用C 1s能谱峰(284.6 eV)进行校准.
1.3 催化剂的活性评价及产物检测
催化剂活性评价在连续流动固定床反应器上进行.不锈钢反应管内径为8 mm,催化剂填装0.1 g,采用石英沙稀释.反应物是含有5.0%萘的癸烷溶液,氢气压力4.0 MPa,温度340℃,反应气液体积比为600.流出产物经空气冷凝收集,反应产物采用7890II气相色谱仪分析,氢火焰离子化检测器(FID),OV-101毛细管色谱柱,柱长50 m,内径0.32 mm.
2 结果与讨论
2.1 催化剂的XRD、TEM、CO化学吸附及XPS表征
为了比较Pd、Pt单金属及不同比例Pd-Pt双金属催化剂的加氢性能,本实验用等体积浸渍法制备了SiO2-Al2O3负载的Pd、Pt单金属催化剂及Pd/Pt比分别为1∶1,1∶4,4∶1的双金属催化剂(简称Pd1Pt1, Pd1Pt4及Pd4Pt1).通过XRD对样品的物相结构进行表征,结果如图1所示.可以看出,无论是Pd、Pt单金属催化剂,还是不同Pd/Pt比双金属催化剂样品均未出现载体SiO2-Al2O3以外的特征衍射峰,这是由于金属在这几种样品上的担载量低,从而使粒子具有较好的分散度.图2为Pd、Pt单金属及不同比例Pd-Pt双金属催化剂的TEM照片,由图可见单金属Pd和Pt催化剂上金属的颗粒较小;Pd1Pt1催化剂表面金属颗粒尺寸大于单金属Pd和Pt;在Pd1Pt4催化剂上发现了尺寸大于10 nm的大颗粒,说明金属在该催化剂上分散得不好,发生了团聚.而Pd4Pt1催化剂上的金属颗粒尺寸最小,约2-3 nm,且粒子分散均匀.

采用CO化学吸附法测定Pd、Pt单金属及不同比例Pd-Pt双金属催化剂的表面金属分散情况,结果如表1所示.可见当Pd与Pt的比例为1∶1或1∶4时,其金属表面分散度及CO化学吸附量均低于单金属Pd和Pt.这可能是由于制备过程中产生了金属的团聚,使暴露在外的活性位相应地减少了.而当Pd与Pt的比例为4∶1时,其金属表面分散度(39.8%)及CO化学吸附量(9.4 μmol·g-1)不仅远高于其他两种比例的双金属催化剂,而且还高于Pd、Pt单金属催化剂,平均粒径(2.8 nm)也是几种样品中最小的.这可能是因为在此种比例的双金属催化剂中,两种金属之间产生的协同效应[15-16]使金属的颗粒更小,并大大提高了活性位在表面的分散度,使其具有更高的活性.


表1 Pd、Pt单金属及不同比例Pd-Pt双金属催化剂金属分散度及CO化学吸附量Table 1 Metal dispersion and CO uptake of monometallic Pd,Pt,and Pd-Pt bimetallic catalysts with different Pd/Pt molar ratios
采用X射线光电子能谱(XPS)测试催化剂的表面价态,结果如图3a所示.所有催化剂样品在335.1 eV处均未能观察到Pd 3d5/2峰,这可能要归因于催化剂的高分散度和低担载量.而在70.1和73.5 eV处,虽然有一个强Al 2p峰的干扰,但通过与纯Pd催化剂的比较,仍能观察到其他样品中归属于Pt0的Pt 4f7/2和 Pt 4f5/2峰的存在 (图 3a,b),这也与Navarro等[17]的研究结果相一致.
2.2 萘在Pd、Pt及Pd4Pt1催化剂上的加氢
为了比较不同种类金属催化剂的加氢性能,我们考察了SiO2-Al2O3担载的Pd、Pt及Pd4Pt1双金属催化剂在340℃,4.0 MPa条件下的萘加氢产物分布随接触时间的变化.
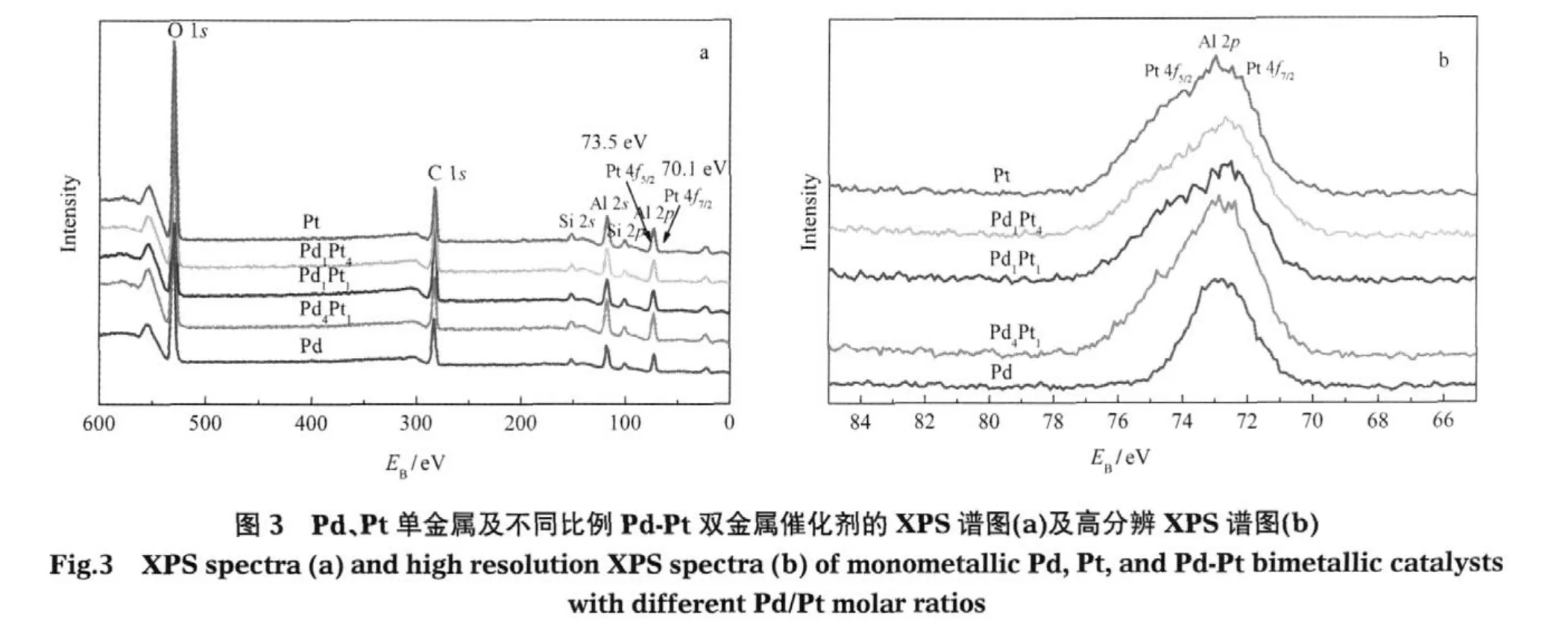

图4显示出随接触时间的增加,三种催化剂上产物的萘含量都呈下降趋势,说明萘的转化率随接触时间的延长而增加.其中萘含量在Pd4Pt1催化剂上的下降最为迅速,其次是在Pd催化剂上,最慢的是在Pt催化剂上.即萘在三种催化剂上的加氢速率顺序为vPd4Pt1>vPd>vPt.产物四氢萘的生成率在三种催化剂上都呈先增高后降低的趋势,在四氢萘的生成率达到最大值并开始降低后,十氢萘的生成率才开始显著增高,说明萘在三种催化剂上的反应都经历了萘→四氢萘→十氢萘的加氢过程.四氢萘的生成率在Pd4Pt1催化剂上达到最大值需要的接触时间最短,并且之后的下降趋势最为明显,说明萘在其上的第一步加氢最快达到平衡,第二步加氢速率也最快,在Pd催化剂上第一步达到平衡较慢,在Pt催化剂上最慢.以产物中的萘含量换算出萘的转化率,以十氢萘(反式+顺式)的选择性(即十氢萘的生成率/萘的转化率)来表示催化剂的深度加氢活性,则在所考察范围内,Pd催化剂上萘的最高转化率为97.5%,对应的十氢萘最高选择性为59.1%,Pt催化剂上萘的最高转化率为96.8%,对应的十氢萘最高选择性为39.9%,而 Pd4Pt1催化剂上萘的最高转化率为98.2%,对应的十氢萘最高选择性为93.6%,远高于两种单金属催化剂.Pd4Pt1催化剂与单金属Pd、Pt催化剂的金属负载量相等,活性却没有介于二者之间,而是高于两种单金属,这说明Pd-Pt双金属中两种金属之间产生了协同效应[15-16].这种效应可能来自于制备过程中Pd、Pt两种金属盐的交互影响,使金属的颗粒更小,分散度提高,比表面积相应增大,从而增加了暴露在催化剂表面的活性位数量[16].这也与之前的表征结果,即Pd4Pt1催化剂表面金属的平均粒径为2.8 nm,小于Pd和Pt催化剂的金属平均粒径(5.7和3.7 nm),Pd4Pt1催化剂的表面金属分散度为39.8%,大于Pd和Pt催化剂的表面金属分散度(19.7%和30.4%)相一致.
四氢萘氢化生成十氢萘过程中实际还有二氢萘和八氢萘生成,但由于其继续氢化的活化能较低,所以很难停留在这两种中间产物上.尤其是1,9-八氢萘,被认为是生成反-、顺-十氢萘最活跃的中间组分[14,18].由于顺-十氢萘热力学上不如反-十氢萘稳定,促使顺-十氢萘向反-十氢萘转化.在负载贵金属的催化剂上,十氢萘还可以通过加氢脱氢来进行立体异构的转化[9],如图5所示.

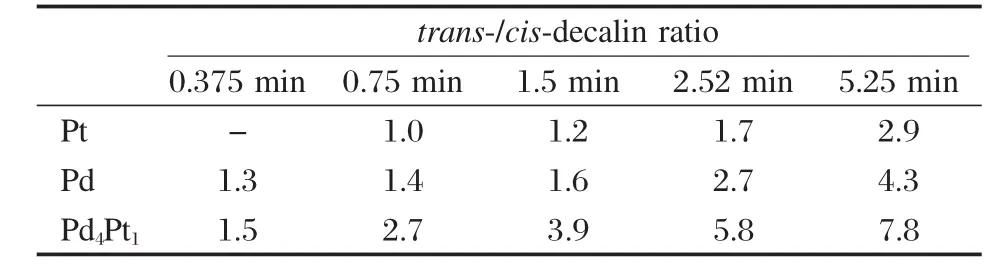
表2 Pd、Pt及Pd4Pt1催化剂在不同接触时间下的十氢萘反/顺生成率之比Table 2 trans-/cis-decalin yield ratio on Pd,Pt,and Pd4Pt1catalysts at different contact time
表2为不同接触时间下Pd4Pt1、Pd和Pt催化剂上的十氢萘反/顺比.在所考察范围内,三种催化剂上的十氢萘反/顺比均随着接触时间的增加而增大.Pd4Pt1上的十氢萘反/顺比最大 (由1.5增至7.8),Pd其次(由1.3增至4.3),均大于1,说明这两种催化剂都倾向于生成反-十氢萘,与之前的文献结论相符[8,13-14].而Pt上的十氢萘反/顺比虽然最小,但也接近1,并未表现出明显的生成顺-十氢萘倾向,而且随接触时间的增大还有少量增加 (由1.0增至2.9).因为Pt没有使十氢萘异构的活性[8],所以这可能是受到了热力学平衡的影响.而相同温度下Pd4Pt1和Pd上的十氢萘反/顺比增幅远大于Pt,说明除了受热力学平衡影响,还有一部分顺-十氢萘在催化剂活性位上异构生成了反-十氢萘.除了接触时间长有利于顺-十氢萘向反-十氢萘转化这一因素,十氢萘的异构还受到竞争吸附的影响.如图6所示,与十氢萘反/顺比升高相伴随的是产物中四氢萘选择性的降低.四氢萘浓度越低越有利于顺-十氢萘向反-十氢萘转化,这是因为四氢萘在活性位上的竞争吸附会强烈抑制十氢萘的异构化反应[19].
2.3 Pd、Pt及Pd4Pt1催化剂的耐硫性能
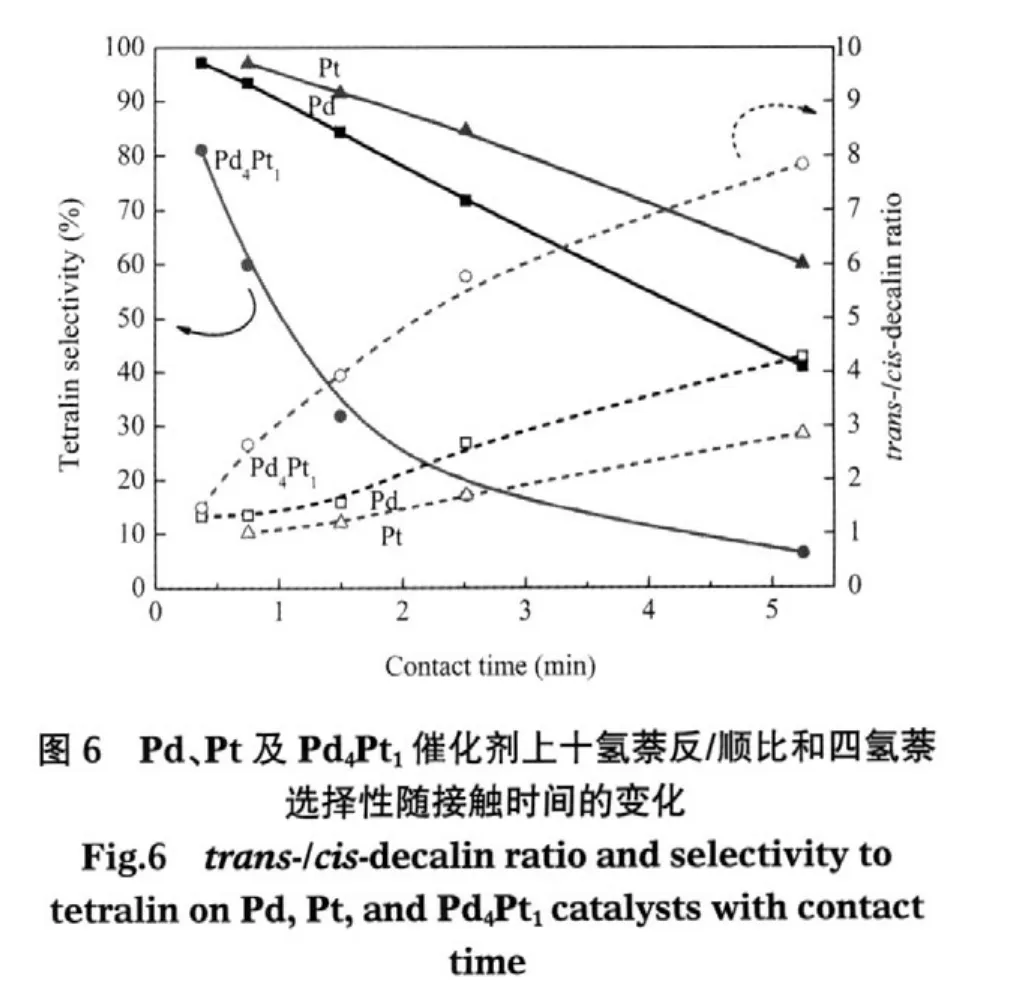
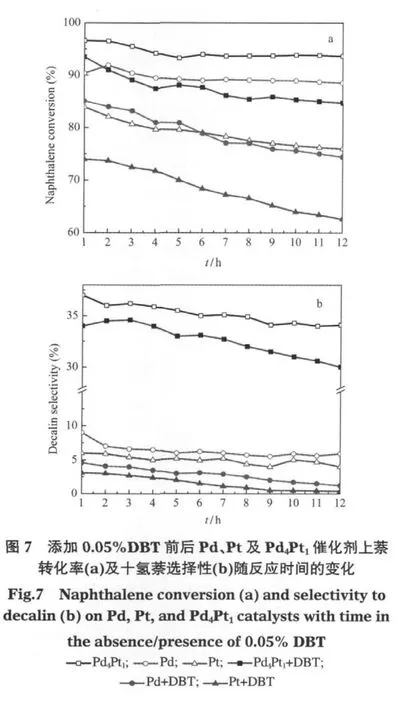
为了比较几种催化剂的耐硫性能,实验选取二苯并噻吩(DBT)作为含硫物质的代表,研究了二苯并噻吩存在下萘在各催化剂上的加氢活性和产物选择性变化.反应条件均为340℃、4.0 MPa、空时0.75 min-1.图7a显示了添加DBT前后Pd、Pt及Pd4Pt1催化剂上萘转化率的变化.在添加DBT之前,Pd4Pt1上的萘转化率最高(95%左右),Pd上的萘转化率其次(约90%左右),Pt上的萘转化率最低.添加DBT后,萘在三种催化剂上的转化率均有降低.Pd4Pt1上的萘转化率仍然高于其他两种单金属,且转化率的降低幅度最小(<10%).而Pd和Pt上的萘转化率降低幅度均大于10%.图7b显示了添加DBT前后Pd、Pt及Pd4Pt1催化剂上的十氢萘选择性变化.在未添加DBT时,Pd4Pt1上的十氢萘选择性就远高于Pd和Pt,添加DBT后,Pd4Pt1上的十氢萘选择性略有降低,而Pd和Pt催化剂上的十氢萘生成几乎完全被抑制.以上现象说明Pd4Pt1催化剂的耐硫性最好,并且即使在有硫存在条件下,饱和第二个芳环的能力也高于另两种单金属.其原因一是Pd4Pt1表面金属的平均粒径小于Pd和Pt催化剂,金属分散度大于Pd和Pt催化剂,具有更大的比表面积,即暴露在外的活性中心,因此具有较高的加氢活性和耐硫性;二是第二种金属的加入改变了原有单一金属的电子性质.许多实验[3-4,20-22]证实在Pd4Pt1催化剂中由于金属间的电子迁移形成了一种不同于Pt-Pt,Pt-Pd金属键的新键,即Ptxδ+—xPdδ-键,在Pd-Pt双金属催化剂中Pt呈缺电子状态,显示正离子性.金属中心的缺电子状态可以抑制硫的不可逆吸附,从而减弱了含硫物种引起的积碳效应[23].
由图8可知添加DBT之前,十氢萘反/顺之比在Pd4Pt1催化剂上最大,在Pd催化剂上次之,在Pt催化剂上最小.这一结果与Jongpatiwut等[18]的结论不同,他们认为Pd-Pt催化剂上的十氢萘反/顺之比接近纯Pd催化剂,从而间接证明之前关于Pd在Pd-Pt双金属催化剂表面富集的报道[22,24-26].这可能是因为在本实验选择的空时(0.75 min-1)下,Pd4Pt1上的四氢萘浓度低于Pd催化剂,所以其上有更多顺-十氢萘在活性位上异构为反-十氢萘.添加DBT后十氢萘反/顺比在Pt催化剂上变化不大,在Pd4Pt1上稍有降低,而在Pd上降低明显.Pt催化剂的活性位不能使十氢萘异构[8],因此十氢萘反/顺比不受中毒影响. Pd4Pt1和Pd催化剂上十氢萘反/顺比因加入DBT而降低,说明原来的反应产物中的确有一部分反-十氢萘是由顺-十氢萘在催化剂活性位上加、脱氢异构生成的,含硫物种在活性位上的吸附阻碍了十氢萘的异构.Pd4Pt1催化剂中Pd与Pt之间的强相互作用使活性位不易与硫结合,因此其十氢萘反/顺比较Pd下降得要少.

2.4 Pd/Pt比对加氢活性及耐硫(DBT)性能的影响
为了研究不同Pd/Pt比对Pd-Pt催化剂加氢活性及耐硫性的影响,分别考察了Pd1Pt1、Pd1Pt4、Pd4Pt1三种催化剂上的萘转化率,十氢萘选择性和反/顺-十氢萘生成率之比在加入DBT前后的变化.反应条件均为340℃、4.0 MPa,空时0.75 min-1.图9a显示未添加DBT时,萘在Pd4Pt1上的转化率最高,在Pd1Pt4上最低.添加DBT(0.05%)后,萘转化率在三种催化剂上均有下降,Pd4Pt1上的萘转化率仍高于另外两种催化剂并且下降幅度最小.添加DBT之前(图9b)十氢萘选择性在Pd4Pt1上最大,在Pd1Pt4催化剂上最小.添加DBT后Pd4Pt1上的十氢萘选择性仍高于另外两种催化剂并且下降幅度最小.Pd4Pt1催化剂的耐硫性最好,一方面是因为Pd4Pt1表面金属平均粒径(2.8 nm)小于Pd1Pt1和Pd1Pt4的金属平均粒径(8.1和15.5 nm),其表面金属分散度(39.8%)大于Pd1Pt1和 Pd1Pt4的表面金属分散度(14.0%和7.2%),因此具有更大的活性金属比表面积,即有更多暴露在外的有效活性中心.另一方面也可能得益于Pd在催化剂表面富集,形成一种Pd—S外壳包裹(保护)Pt核的结构[22].电子从Pt到Pd的迁移能够增强Pd—S键的强度,而使Pt不易与硫结合.
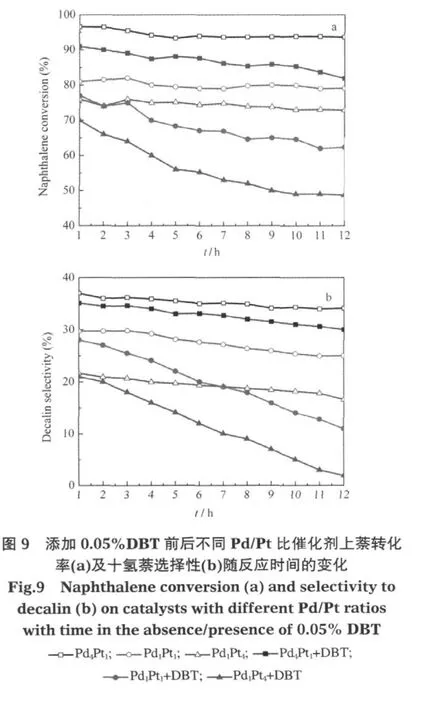
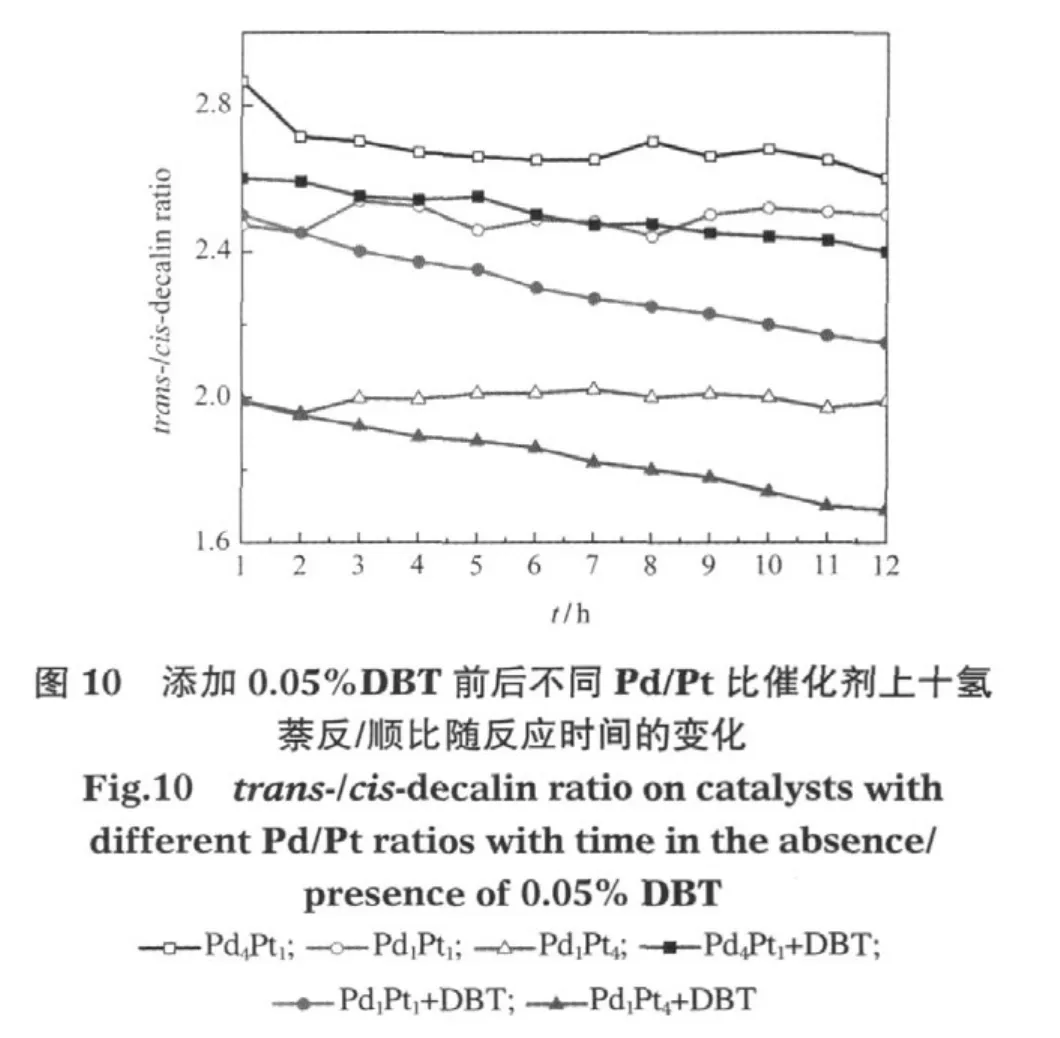
图10显示,添加DBT之前Pd4Pt1催化剂上反/顺-十氢萘之比最大,Pd1Pt4催化剂上反/顺-十氢萘之比最小.添加DBT后,Pd4Pt1催化剂上十氢萘反/顺比稍有降低,Pd1Pt4和Pd1Pt1催化剂上的十氢萘反/顺比降低较明显,同样说明原来的反应产物中有一部分反-十氢萘是由顺-十氢萘在催化剂活性位上加、脱氢异构生成的.DBT的加入阻止了部分顺-十氢萘向反-十氢萘的异构.
3 结 论
制备了SiO2-Al2O3负载的Pd、Pt单金属催化剂及不同Pd/Pt比的Pd-Pt双金属催化剂,考察了各催化剂的萘加氢活性、生成的产物分布以及硫对上述性质产生的影响.实验结果表明,Pd4Pt1催化剂无论是加氢速率、萘转化率、产物十氢萘选择性还是耐硫性,都优于单金属催化剂.其上的十氢萘反/顺比也更大.在所考察范围内,Pd4Pt1催化剂上的萘转化率最高可达98.2%,十氢萘选择性可达93.6%.添加DBT后Pt催化剂上十氢萘反/顺比不受影响,Pd催化剂上的十氢萘反/顺比降低较Pd4Pt1催化剂明显,说明原来的反应产物中有一部分反-十氢萘是由顺-十氢萘在催化剂活性位上加、脱氢异构生成的.在Pd/Pt比分别为 4∶1、1∶1、1∶4的双金属催化剂中, Pd4Pt1催化剂的活性和十氢萘选择性最高,耐硫性能也最佳.
1 Lee,S.L.;Wind,M.D.;Desai,P.H.;Johnson,C.C.;Mehmet,Y. A.Fuel Reform.,1993,5:26
2 Khan,M.R.;Reynolds,G.Chemtech,1996,26:56
3 Yasuda,H.;Yoshimura,Y.Catal.Lett.,1997,46:43
4 Koussathana,M.;Vamvouka,D.;Economou,H.;Verykios,X. Appl.Catal.,1991,77:283
5 Fujikawa,T.;Idei,K.;Ebihara,T.;Mizuguchi,H.;Usui,K.Appl. Catal.A,2000,192:253
6 Lin,T.B.;Jan,C.A.;Chang,J.R.Ind.Eng.Chem.Res.,1995,34: 4284
7 Zhu,H.Y.;Zhang,Y.;Qiu,Z.G.;Cui,H.T.;Zhao,L.F.Fine Chem.,2009,26:5 [朱红英,张 晔,邱泽刚,崔海涛,赵亮富.精细化工,2009,26:5]
8 Schmitz,A.D.;Bowers,G.;Song,C.Catal.Today,1996,31:45
9 Lai,W.C.;Song,C.Catal.Taday,1996,31:171
10 Kumata,F.;Hirasawa,Y.Method for producing decalin from naphthalene by two stage hydrogeneration reaction:Japan, JP2003160515[P].2003-06-03
11 Kumata,F.;Hirasawa,Y.Method for producing decalin by hydrogenating naphthalene:Japan,JP2003212800[P].2003-07-30
12 Jaffe,F.Preparation of cis-decalin:US,3349139[P].1967-10-24
13 Shoichiro,M.;Masakazu,H.Hydrogenation catalyst:Japan, JP51121495[P].1976-10-23
14 Weitkamp,A.W.Adv.Catal.,1968,18:l
15 Matsui,T.;Harada,M.;Bando,K.K.;Toba,M.;Yoshimura,Y. Appl.Catal.A,2005,290:73
16 Niquille-Röthlisberger,A.;Prins,R.J.Catal.,2006,242:207
17 Navarro,R.M.;Pawelec,B.;Trejo,J.M.;Mariscal,R.;Fierro,J.L. G.J.Catal.,2000,189:184
18 Jongpatiwut,S.;Li,Z.R.;Resasco,D.E.;Alvarez,W.E.;Sughrue, E.L.;Dodwell,G.W.Appl.Catal.A,2004,262:241
19 Huang,T.C.;Kang,B.C.Ind.Eng.Chem.Res.,1995,34:1140
20 Matsubayashi,N.;Yasuda,H.;Imaura,M.;Yoshimura,Y.Catal. Today,1998,45:375
21 Yasuda,H.;Matsubayashi,N.;Sato,T.;Yoshimura,Y.Catal.Lett., 1998,54:23
22 Guillon,E.;Lynch,J.;Uzio,D.;Didillon,B.Catal.Today,2001, 65:201
23 Lee,J.K.;Rhee,H.K.J.Catal.,1998,177:208
24 Fujikawa,T.;Tsuji,K.;Mizuguchi,H.;Godo,H.;Idei,K.;Usui,K. Catal.Lett.,1999,63:27
25 Hansen,P.L.;Molenbroek,A.M.;Ruban,A.V.J.Phys.Chem., 1997,101:1861
26 Fiermans,L.;De Gryse,R.;De Doncker,G.;Jacobs,P.A.; Martens,J.A.J.Catal.,2000,193:108
Naphthalene Hydrogenation Activity over Pd,Pt and Pd-Pt Catalysts and Their Sulfur Tolerance
ZHANG Xiao-Fei SHAO Zheng-Feng MAO Guo-Qiang HE De-Min ZHANG Qiu-Min*LIANG Chang-Hai*
(State Key Laboratory of Fine Chemicals,School of Chemical Engineering,Dalian University of Technology,Dalian 116012, Liaoning Province,P.R.China)
Monometallic Pd and Pt as well as bimetallic Pd-Pt catalysts(Pd1Pt1,Pd1Pt4,and Pd4Pt1)with Pd/Pt molar ratios of 1∶1,1∶4,and 4∶1 supported on SiO2-Al2O3were prepared by incipient-wetness impregnation and characterized by X-ray diffraction(XRD),transmission electron microscopy(TEM),CO chemisorption,and X-ray photoelectron spectroscopy(XPS).Their catalytic activities toward naphthalene hydrogenation and the sulfur tolerance of these catalysts were investigated.We found that naphthalene conversion,selectivity toward decalin,and the trans-/cisdecalin yield ratio on Pd4Pt1were 98.2%,93.6%,and 7.8,respectively,which are higher than those on Pd(97.5%,59.1%, 4.3)and Pt(96.8%,39.9%,2.9).The rate of naphthalene hydrogenation on the three catalysts increased according to: vPd4Pt1>vPd>vPt.In the presence of dibenzothiophene(DBT),the naphthalene conversion and selectivity toward decalin for Pd4Pt1were still the highest.The trans-/cis-decalin ratio was not affected on the Pt catalyst,but it did decrease slightly on Pd4Pt1and it decreased observably on the Pd catalyst.The Pd4Pt1catalyst also presented the highest naphthalene conversion and selectivity toward decalin among the three bimetallic catalysts studied in the presence and absence of DBT.
Noble metal;Naphthalene;Hydrogenation;Sulfur tolerance; trans-/cis-decalin yield ratio
O643
Received:May 6,2010;Revised:July 21,2010;Published on Web:August 24,2010.
*Corresponding authors.ZHANG Qiu-Min,Email:qmzhang@chem.dlut.edu.cn;Tel:+86-411-39893865.
LIANG Chang-Hai,Email:changhai@dlut.edu.cn;Tel:+86-411-39608806.
The project was supported by the National Natural Science Foundation of China(20973029)and Fundamental Research Funds for the Central Universities,China.
国家自然科学基金(20973029)和中央高校基本科研业务费专项资金资助项目.
ⒸEditorial office of Acta Physico-Chimica Sinica
猜你喜欢
机械工业标准化与质量(2022年6期)2022-08-12
航天工业管理(2020年9期)2020-12-28
重型机械(2020年2期)2020-07-24
热力透平(2020年2期)2020-06-22
石油化工建设(2018年2期)2018-07-11
今日自动化(2018年4期)2018-05-06
中国调味品(2017年2期)2017-03-20
中国石油大学学报(自然科学版)(2015年2期)2015-11-10
橡胶工业(2015年10期)2015-08-01
中学化学(2015年2期)2015-06-05
