
三苯氧基三对硝基苯氧基环三磷腈的合成与表征
2010-09-14 10:21:24丁炎可徐龙鹤
沈阳化工大学学报 2010年4期
丁炎可, 徐龙鹤
(1.沈阳化工大学应用化学学院,辽宁沈阳 110142; 2.沈阳化工研究院,辽宁沈阳 110021)
三苯氧基三对硝基苯氧基环三磷腈的合成与表征
丁炎可1, 徐龙鹤2
(1.沈阳化工大学应用化学学院,辽宁沈阳 110142; 2.沈阳化工研究院,辽宁沈阳 110021)
以苯酚钠、对硝基苯酚钠和六氯环三磷腈 (HCCTP)为原料合成三苯氧基三对硝基苯氧基环三磷腈,并采用 FT-IR、1H-NMR、LC-MS、13C-NMR和31P-NMR对产物进行表征.以四氢呋喃为溶剂,采用摩尔比为 1∶3∶3.5的 HCCTP、苯酚钠和对硝基苯酚钠,反应温度 65℃,反应时间 7 h,得到三苯氧基三对硝基苯氧基环三磷腈.
六氯环三磷腈; 三苯氧基三对硝基苯氧基环三磷腈; 合成
磷腈类化合物是一类骨架由磷氮单双键交替排列而成的化合物,其独特的杂环结构和较高的磷氮含量使之具有良好的热稳定性和阻燃性[1-4].六氯环三磷腈 (HCCTP)的合成[5-6]及其与酚类[7-8]的反应研究较多,但对同一磷原子上引入不同的化合物可以扩大磷腈类化合物的应用范围,并且这类化合物合成报道较少[9-10].本文以六氯环三磷腈、苯酚钠和对硝基苯酚钠为原料合成了三苯氧基三对硝基苯氧基环三磷腈,用液相色谱对反应进程进行跟踪,对目标化合物进行分离和结构鉴定.
1 实验部分
1.1 原料与试剂
六氯环三磷腈,山东蓝印化工;苯酚钠,自制[11];对硝基苯酚钠,自制[11];四氢呋喃,分析纯,国药试剂;甲苯,分析纯,国药试剂.
1.2 三苯氧基三对硝基苯氧基环三磷腈的合成
向装有温度计、冷凝管、磁力搅拌器的 50 mL三口瓶中依次投入 5 g(0.014 mol)六氯环三磷腈(化合物 1)和 20 mL四氢呋喃,常温下搅拌溶解.向反应瓶中滴入 1.66 g(0.014 mol)苯酚钠的 6 mL四氢呋喃溶液,用时 0.5 h,滴毕后将反应液加热到50℃保温反应 1.5 h,苯酚钠反应完全,降温到 30℃.继续向反应瓶中滴入1.66 g(0.014 mol)苯酚钠的 6 mL四氢呋喃溶液,用时 0.5 h,滴毕后将反应液加热到 50℃保温反应 1.5 h,苯酚钠反应完全,降温到 30℃.继续向反应瓶中滴入 1.66 g(0.014 mol)苯酚钠的 6 mL四氢呋喃溶液,用时 0.5 h.加热至回流反应 2 h,苯酚钠反应完全.向反应瓶中加入 8.1 g(0.05 mol)对硝基苯酚钠,回流反应 3 h,降温冷却.反应液脱溶后,加入 30 mL蒸馏水搅拌过滤,所得固体进行柱层析 (V(EA)∶V(PE)= 1∶9),可得到目标产物三苯氧基三对硝基苯氧基环三磷腈 (化合物 3)的白色晶体 7.5 g.mp:129~131℃.其合成路线如图 1所示.


图 1 三苯氧基三对硝基苯氧基环三磷腈的合成路线Fig.1 Synthesis route of tri-phenoxy-tri-(4-nitrophenoxy)-cyclotriphosphazene
1.3 测试与仪器
傅里叶变换红外光谱 (FT-IR)采用美国Nicolet公司的 Nicolet Impact 400型;核磁共振(NMR)采用美国 Varian公司的 Varian Mercury V×300核磁共振谱仪;液相色谱-质谱联用仪采用美国Agilent公司的AgilentLC/MSD Trap VL; HPLC由Agilent 1100高效液相色谱仪测定;熔点测定仪为 RY-Ⅰ型.
1.4 液相色谱条件
色谱柱:ODS-35μm C18柱 (4.6 mm×250 mm);流动相:乙腈和水;流速:1.0 mL/min;柱温:室温;检测波长:225 nm.
2 结果与讨论
2.1 反应液跟踪检测
为考察反应物 1中 2个氯的反应活性,得到被苯酚均三取代的中间体 2,然后再引入硝基酚来合成目标化合物 3,实验过程中将苯酚钠分 3次加入,每次加入 1份 (每份与化合物 1的摩尔比为 1∶1).每次加料完毕反应 1.5 h,然后进行液谱跟踪反应,3次的跟踪结果如图 2所示.图2(a)、图 2(b)分别为加入 1份、2份苯酚钠后的液谱数据 (流动相:V(乙腈)/V(水)=90/10). 2.9 min处吸收峰为原料苯酚钠.对比图 2(a)和图 2(b)可以发现:加入第 2份苯酚钠后,随着反应的进行,11.2 min处的吸收峰减小,12.2 min处的吸收峰明显增大.据此推测 11.2 min和12.2 min处的吸收峰分别为化合物 1的苯酚单取代和二取代产物.
图 2(c)为加入第 3份苯酚钠后的液谱数据(流动相 ∶V(乙腈)/V(水)=85/15).采用图2(a)、图 2(b)的液谱条件无法对第 3次样品进行有效分离,因此对液谱条件进行了调整.通过对比发现:图 2(c)中 18.3 min处的吸收峰为图2(b)中 12.2 min处的吸收峰,信号强度明显减小,并出现了 19.7 min处较强的吸收峰.据此推测该峰为化合物 1的苯酚均三取代产物.

图 2 液谱跟踪反应的数据Fig.2 The data of HPLC traced reaction
2.2 产物 3的31P-NMR分析
图 3为产物 3的31P-NMR图.图 3中δ= 0.000处是基准物H3PO4的峰,δ=7.940处是产物 3的峰.从图 3可以看出产物具有单一的峰,说明产物 3中所有的磷原子都处于相同的化学环境.

图 3 三苯氧基三对硝基苯氧基环三磷腈的31P-NMR图Fig.331P-NMR of tri-phenoxy-tri-(4-nitrophenoxy)-cyclotriphosphazene
2.3 产物3的MS分析
图 4为产物 3的 MS图.峰值 829与产物 3的理论相对分子质量 (828)加氢 (1)的值一致,并且 851处的峰值符合产物 3的理论相对分子质量 (828)加钠(23)的值.

图 4 三苯氧基三对硝基苯氧基环三磷腈的MS图Fig.4 MS of tri-phenoxy-tri-(4-nitrophenoxy)-cyclotriphosphazene
2.4 产物 3的 FT-IR分析
从图 5可以看到:1 268 cm-1、1 178 m-1处出现了环三磷腈的 P==N伸缩振动吸收峰, 3 078 cm-1为苯环上的 C—H振动吸收峰;1 488 cm-1和 1 590 cm-1是苯环的骨架变形振动吸收峰,表明产物结构中存在磷腈杂环和苯环;1 519 cm-1和 1 347 cm-1分别为硝基的不对称和对称伸缩振动吸收峰;1 109 cm-1、1 024 cm-1和 945 cm-1峰为 P—O—C的特征吸收峰;602 cm-1处的 P—Cl吸收峰完全消失,说明 HCCTP磷上的氯原子已被苯酚和对硝基酚完全取代.

图 5 三苯氧基三对硝基苯氧基环三磷腈的 IR图Fig.5 IR of tri-phenoxy-tri-(4-nitrophenoxy)-cyclotriphosphazene
2.5 产物3的13C-NMR分析
图 6为产物 3的13C-NMR谱图.谱图中δ= 76.5~77.4为溶剂氯仿峰,δ=120.6~129.7代表苯环中 5种叔碳的峰,δ=144.8~155.0代表苯环中 3种季碳的峰,与产物 3有相同的结构特征.

图 6 三苯氧基三对硝基苯氧基环三磷腈的13C-NMR图Fig.613C-NMR of tri-phenoxy-tri-(4-nitrophenoxy)-cyclotriphosphazene
2.6 产物3的1H-NMR分析
硝基邻位的 H因为有硝基的强吸电子作用,使邻位 H偏向低场,与其他 H的化学位移相差较大,所以根据这个性质也得到了一个判断各取代集团取代数量的有效信息,表 1为不同取代集团个数与积分比例的关系.从表 1可以清晰地看到随取代基比例的不同,不同区域的积分面积有很大的变化,这也是判断化合物结构的一个有效手段.

表 1 不同取代比例的影响Table 1 The effect of different substituent ratio
图 7为产物 3的1H-NMR(300 MHz, DMSO):δ=8.02~8.11(m,硝基邻位 H),6.92~7.27(m,其他苯环 H)积分面积比为 1∶3.5,没有酚 H的吸收,2.5(溶剂峰),3.07(水峰).
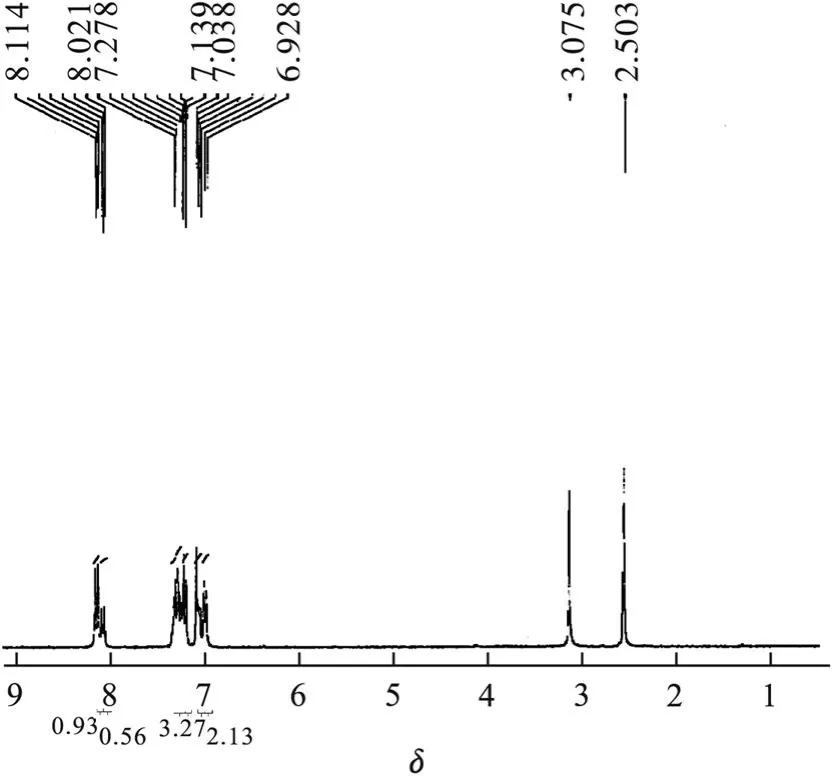
图 7 三苯氧基三对硝基苯氧基环三磷腈的1H-NMR图Fig.71H-NMR of tri-phenoxy-tri-(4-nitrophenoxy)-cyclotriphosphazene
3 结 论
(1)以苯酚钠、对硝基苯酚钠和 HCCTP为原料,在 THF中回流反应,经柱层析分离得到目标化合物三苯氧基三对硝基苯氧基环三磷腈.
(2)采用 FT-I R、1H-NMR、LC-MS、13C-NMR和31P-NMR对产物进行了表征.
致谢:文章在博美达有限公司的资助下完成,在此表示感谢.
[1] Choi Seong-w oo,O hba Sharon,B runovska Zdenka, et al.Synthesis Characterization and Therm alDegradation of Functional Benzoxazine M onom ers and Polym ers Containing Phenylphosphine O xide[J]. Polym er D egradation and Stability,2006,91(5): 1166-1178.
[2] 邵宗龙,胡源,何前锋,等.聚磷嗪化合物的合成及其阻燃性能的研究[J].合肥工业大学学报 (自然科学版),2000,23(4):601-603.
[3] 大冢化学株式会社.交联的苯氧基膦腈化合物、其制备方法、阻燃剂、阻燃性树脂组合物和阻燃性树脂成形体:中国,CN99809510[P].1999-01-09.
[4] Z im a V,Svoboda J,Benes L,et al.Synthesis and Characterization of New Strontium4-carboxy phenylphosphonates[J].Journal of Solid State Chem istry, 2007,180(3):929-939.
[5] Fukuoka N aohiko,Yasuda Heinosuke,N ish im atsu M asayuki,et al.Phosphazene Composition Fam e Retardant Containing the Sam e and Production M ethod Therefor:JP,2002322188[P].2002-11-08.
[6] Fujita Tatsuya,Fujio Tsuyom u.Im age Reader:JP, 02252762[P].2002-09-06.
[7] W ang Pin-sheng,Chiu W en-yen,Chen Leo-w ang, et al.Therm alD egradation Behavior and Flamm ability of Polyurethanes B lended with Poly(B ispropoxyphosphazene)[J].Polym erD egradation and Stability,1999,66(3):307-315.
[8] 孙启新,王利生.环状六氯环三磷腈的合成[J].化工新型材料,2003,31(10):18-20.
[9] Kum arD edra,Stclair Erry L.Process or Preparing is Om eric Trisaryloxycylotriphosphazene Polym er Precursors and Interm ediate:WO,8912639[P].1989-12-18.
[10]U eyam a Shinichiro.Process for Producing A ryloxysubstituted Phosphazene D erivatives: USA, US5075453[P].1991-12-24.
[11]陈长明.精细化学品制备手册[M].北京:企业管理出版社,2004:45-46.
Synthesis and Characterization of Tri-phenoxy-tri-(4-nitrophenoxy)-cyclotriphosphazene
D ING Yan-ke1, XU Long-he2
(1.Shenyang U niversity of Chem ical Technology,Shenyang 110142,China; 2.Shenyang Research Institute of Chem ical Industry,Shenyang110012,China)
Tri-phenoxy-tri-(4-nitrophenoxy)-cyclotriphos phazene was synthesized by using sodium phenolate,sodium 4-nitrophenolate and hexachlorocy clotriphosphazene as raw m aterials.The product was characterized by FT-IR,1H-NM R,LC-M S,13C-NM R and31P-NM R.HCCTP,sodium phenolate and sodium4-nitrophenolate taken in the m olar ratio of 1∶3∶3.5 were dissolved in tetrahydrofuran (THF),reacting at 65℃ for 7h and the product was gained.
hexachlorocy clotriphosphazene; tri-phenoxy-tri-(4-nitrophenoxy)-cyclotriphosphazene; synthesis
TQ126.2
A
1004-4639(2010)04-0303-05
2010-01-04
丁炎可(1983-),男,河南郑州人,硕士研究生在读,主要从事磷腈阻燃剂方面的研究.
徐龙鹤(1955-),朝鲜族,男,辽宁沈阳人,教授级高级工程师,博士,主要从事精细化工方面的研究.
猜你喜欢
云南化工(2020年11期)2021-01-14 00:50:54
铜仁学院学报(2018年6期)2018-07-05 09:47:36
中国洗涤用品工业(2016年2期)2016-02-28 19:03:18
合成化学(2015年4期)2016-01-17 09:01:27
中国塑料(2015年2期)2015-10-14 05:34:31
云南中医学院学报(2015年2期)2015-07-31 18:11:59
应用化工(2014年1期)2014-08-16 13:34:08
应用化工(2014年4期)2014-08-16 13:23:09
中国氯碱(2014年10期)2014-02-28 01:04:59
化工生产与技术(2014年6期)2014-02-27 13:42:07
