
G蛋白偶联受体及其类型的预测
2010-09-13 06:04吴建盛马昕周童汤丽华胡栋
Biophysics Reports 2010年2期
吴建盛,马昕,周童,汤丽华,胡栋
1.南京邮电大学地理与生物信息学院,南京210046;
2.东南大学生物电子学国家重点实验室,南京210096
G蛋白偶联受体及其类型的预测
吴建盛1,马昕2,周童2,汤丽华1,胡栋1
1.南京邮电大学地理与生物信息学院,南京210046;
2.东南大学生物电子学国家重点实验室,南京210096
G蛋白偶联受体是非常重要的信号分子受体,其功能失调会导致许多疾病的产生。在前期工作的基础上,作者将序列特征分析与支持向量机技术结合起来,通过分析序列的特征差异,对G蛋白偶联受体分子及其类型进行识别。首次提取了G蛋白偶联受体对应的mRNA序列的绝对密码子使用频率作为特征,这主要因为它既包含了基因密码子使用偏性的信息,也包含了基因所编码蛋白的氨基酸组成信息。结果显示:在G蛋白偶联受体序列及其类型预测的问题中,设计支持向量机分类器时,最好选择使用包含基因序列绝对密码子使用频率和蛋白序列双联氨基酸使用频率两部分信息的组合特征作为特征,同时采用径向基核作为核函数。
G蛋白偶联受体;支持向量机;绝对密码子使用频率
0 引言
G蛋白偶联受体(G-protein coupled receptor,GPCR)是一类具有7个跨膜螺旋的跨膜蛋白受体,能结合并调节G蛋白活性,是一类非常重要的信号分子受体。GPCR的结构特征及其在信号传导中的重要作用,决定了其可以作为重要的药物靶点。GPCR的功能失调会导致许多疾病的发生,如阿尔茨海默氏症、帕金森症、侏儒症、色盲症、色素性视网膜炎和哮喘等。通过调节有关GPCR介导的信号传导,可以治疗抑郁症、精神分裂症、失眠、高血压、虚弱、焦躁、紧张、肾功能衰竭、心脑血管疾病和炎症等病症。大部分药物可通过靶向作用于GPCR而达到治疗的效果,所以GPCR在制药领域中占有极其重要的地位。根据GPCR的序列差异,GPCR蛋白超家族可分为5类,准确地分类预测GPCR有着很重要的意义和作用。
G蛋白偶联受体是重要的药物靶标,很多药物方面的研究瞄准它们的结构与功能的关系[1]。然而,大多数GPCR的三级结构仍然是未知的,主要是由于这些蛋白难于结晶。同时,这些蛋白在一般的溶剂中溶解度都不大,使得核磁共振也无法使用。相反,随着人类基因组以及其它种类生物基因组计划的开展,已经获得了大量的氨基酸序列数据。目前,如何利用这些已知的一级结构信息,成为生物信息学的研究热点之一,比如,如何从大量蛋白质序列中找出GPCR,找到GPCR后,又如何判断它的类型等。
近年来,许多识别算法已经应用于GPCR类型的预测,如利用BLAST在数据库中搜索相似序列[2]、基于氨基酸物理化学特性的统计方法[3]、基于进化树[4]以及基于隐马尔科夫链[5]等方法。上述方法主要依赖于序列间的相似性,当要判断的序列与训练集样本间缺乏相似性时,预测结果受到限制;同时,这些方法大多基于传统的统计理论,对训练集样本的数目有一定要求,而现有的已知类别样本有限,这同样影响了分类预测的准确率。针对这种情况,Karchin等[6]开始尝试利用支持向量机(support vector classification,SVM)的方法来识别GPCR超家族中各蛋白的类型,并取得了一定的效果。特别是,Bhasin等[7]在Karchin等思路的基础上,还是利用支持向量机,并结合蛋白质一维序列的双联氨基酸使用频率,对GPCR的蛋白质类型进行预测,得到了很好的效果。
然而,上述的方法基本都是基于氨基酸序列的特征。目前,还很少有直接从编码GPCR蛋白的核酸序列中提取特征进行GPCR蛋白类型预测的方法。本文中,我们基于前期研究工作的基础[8~10],将序列特征分析与支持向量机结合起来,首次提取GPCR蛋白对应的mRNA基因序列中的绝对密码子使用频率信息,并加入蛋白质双联氨基酸使用频率信息,对GPCR蛋白序列及其类型进行识别,取得了很好的效果,并且与基于单联氨基酸使用频率的方法,以及目前预测效果最好的Bhasin等的基于双联氨基酸使用频率的方法[7]进行了比较。
1 数据及方法
1.1 GPCR数据来源
GPCRDB数据库(http://www.gpcr.org/7tm/)[11]是一个专注于收集、整合G蛋白偶联受体(GPCR)信息的数据库,其中的GPCR蛋白序列数据主要来源于SWISS-PROT数据库。根据GPCRDB数据库,GPCR蛋白共分为5大类,与Structural Classification of Proteins (SCOP,http://www.bio.cam.ac.uk/scop/)数据库中的分类一致。本文中,我们收集了SWISS-PROT数据库中的GPCR蛋白序列数据,其中,A类序列690个,B类序列142个,C类序列240个,D类序列655个,E类序列37个。为了衡量分类器对GPCR序列识别的效果,我们增加了99个非GPCR的欺骗序列(decoy),这些序列来源于Karchi等[6]的实验。
同时,为了从核酸的角度提取序列特征,我们利用GPCR序列在SWISS-PROT的注释信息,编写了perl程序,从EMBL-EBI(http://srs.ebi.ac.uk/srsbin/cgi-bin/wgetz?-page+srsq2+-noSession)获得了每个GPCR蛋白对应的mRNA序列。
1.2 序列特征的提取
对于GPCR的分类问题,本文提取序列特征的方法主要有三种:1)蛋白序列的单联氨基酸使用频率(single amino acid use frequency);2)为了和目前预测效果最好的Bhasin等的方法[7]进行比较,我们提取了蛋白序列的双联氨基酸使用频率信息;3)包含基因序列的绝对密码子使用频率和蛋白序列的双联氨基酸使用频率两部分信息的组合特征(hybridfeature)。
单联氨基酸使用频率(Fi)是氨基酸i在该段蛋白序列中的出现频率,其计算方法如式(1),其中,n指整段蛋白序列中氨基酸的个数,Ai是氨基酸i在该段蛋白序列中出现的次数。对于每条蛋白序列提取该特征,可转化为一个20维的数字向量(20种氨基酸),向量的每个元素对应一种氨基酸在该蛋白序列中出现的频率。

双联氨基酸使用频率(Fij)是双联氨基酸i和j在该段蛋白序列中的共同出现频率,其计算方法如式(2),其中,m指整段蛋白序列中双联氨基酸的个数。Aij是双联氨基酸ij在该段蛋白序列中出现的次数。每条蛋白序列可转化为一个400维的数字向量,向量的每个元素对应一种双联氨基酸在蛋白序列中出现的频率。

在本文中,我们提取GPCR蛋白对应的mRNA序列的每种密码子的绝对密码子使用频率(codon use frequency,FCU),作为密码子使用偏性的衡量标准,它的计算公式如下:

其中,obsi指某一特定的密码子i在基因中出现的次数;total指整段基因中的密码子的个数。这种衡量方法的优势在于,它含有较多的序列信息。首先,它包含了基因的密码子使用偏性信息。其次,它还含有基因所编码蛋白的氨基酸组成信息。每个GPCR蛋白样本可转化为一个64维的数字向量,向量的每个元素代表一种密码子在GPCR蛋白对应的mRNA序列中出现的绝对密码子使用频率。这样,本文中使用的包含基因序列的绝对密码子使用频率和蛋白序列的双联氨基酸使用频率两部分信息的组合特征,为一个464维的数字向量。
1.3 支持向量机
支持向量机(SVM)是Vapnik等[12]提出的一类新型机器学习方法。由于其出色的学习性能,在高维小训练样本情况下有着很好的泛化能力,该技术已成为机器学习界的研究热点,并在很多领域都得到了成功应用。它是以结构化风险最小化(structural risk minimization,SRM)代替常用的经验风险最小化(empirical risk minimization,ERM)作为优化准则,其基本思想是,对于非线性可分样本,将其输入向量经非线性变换映射到另一个高维空间Z中,在变换后的空间中寻找一个最优的分界面(超平面),使其推广能力最好。具体应用SVM的步骤为:1)选择适当的核函数;2)求解优化方程,获得支持向量及相应的Lagrange算子;3)写出最优分界面方程。在本文中,为了实现SVM算法,我们采用了R语言的e1071软件包(version 1.5-16)[13]。
1.4 模型性能评价
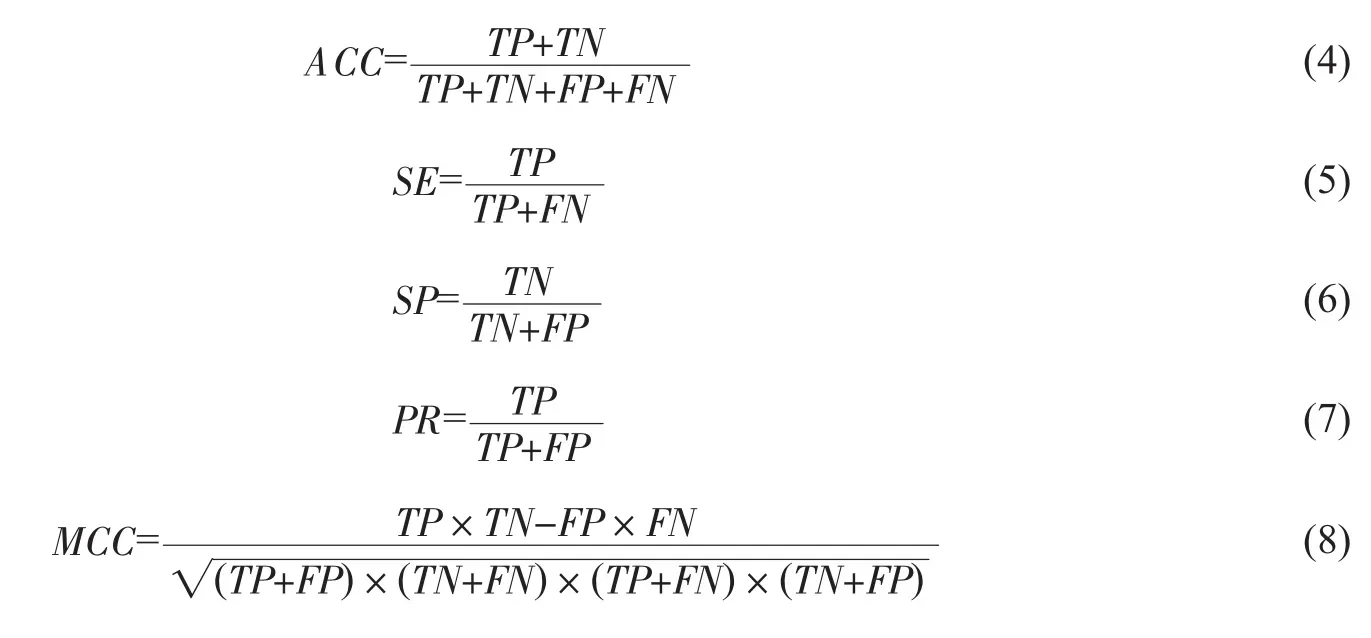
对于分类预测问题,所预测的样本有4种情况:假阳性(false positive,FP),真阳性(true positive,TP),假阴性(false negative,FN),真阴性(true negative,TN)。其总体预测准确率(accuracy,ACC),特异性(specificity,SP),敏感性(sensitivity,SE)和Mattew相关系数(Mattew’s correlation coefficient,MCC)[14]的定义如下:
1.5 实验流程
本文中,对GPCR的分类可分为两步来操作:第一步是用SVM从蛋白序列集中找出GPCR序列;第二步,对识别出的GPCR序列进一步分类,确定其所属的类别,共涉及6个SVM分类器。在第一步的GPCR序列识别中,我们把A、B、C、D、E 5类共1764条GPCR序列合并,作为机器学习的正类集,99条非GPCR的欺骗序列(decoy)作为机器学习的负类集。首先根据1.2节描述的特征提取的方法,将蛋白序列转换为可供SVM软件识别使用的数字向量序列,然后使用十倍交叉验证(ten-fold cross-validation)的方法来衡量分类器的性能。所谓十倍交叉验证是指,利用随机数抽取的方法,将数据集随机分成数量相等的10个数据集,将其中9个数据集作为训练集,剩下的一个作为测试集,通过分类器来进行分类预测,然后重新分配训练集与测试集,重复刚才的过程,如此这般,一共需要作10次训练及测试,利用这10次实验的结果来衡量分类器的性能。第二步中,我们需要对GPCR超家族在类别层次上进行分类预测。这是一个多类的分类问题,我们可以将此多类问题转化为两类问题。设计5个SVM分类器。当对A类进行分类预测时,将A类样本作为机器学习的正类集,其余4类合并作为负类集,通过SVM来进行分类预测。对于GPCR的其余类别,方法类似。通过对这5个分类器所有输出结果的分析,得出最终的分类结果。
2 实验结果
2.1 GPCR序列的识别
首先要做的是,测试我们的SVM模型从众多序列中识别出GPCR的能力。我们将A、B、C、D和E 5类GPCR共1764条序列合并为正类集,然后以99个欺骗序列作为负类集,进行十倍交叉验证,结果如表1所示。
表1中第一列核函数是在SVM学习过程中所采用的核函数类别,包括线性核函数、多项式核函数和径向基核函数。第二列的特征指出了从序列中抽取特征时所采用的方法,包括单联氨基酸频率、Bhasin等的双联氨基酸使用频率,以及我们提出的Hybrid feature。对于多项式核函数,我们设定参数cost=1.0,并采用3阶多项式核;对于径向基核函数,我们还是设定参数cost=1.0,而参数gamma在单联氨基酸频率、双联氨基酸使用频率及Hybrid feature中分别取1/20、1/400和1/464。从表1中可以看出,使用各种核函数及利用3种序列特征都取得了非常好的结果,预测准确率(ACC)都在98.50%以上。
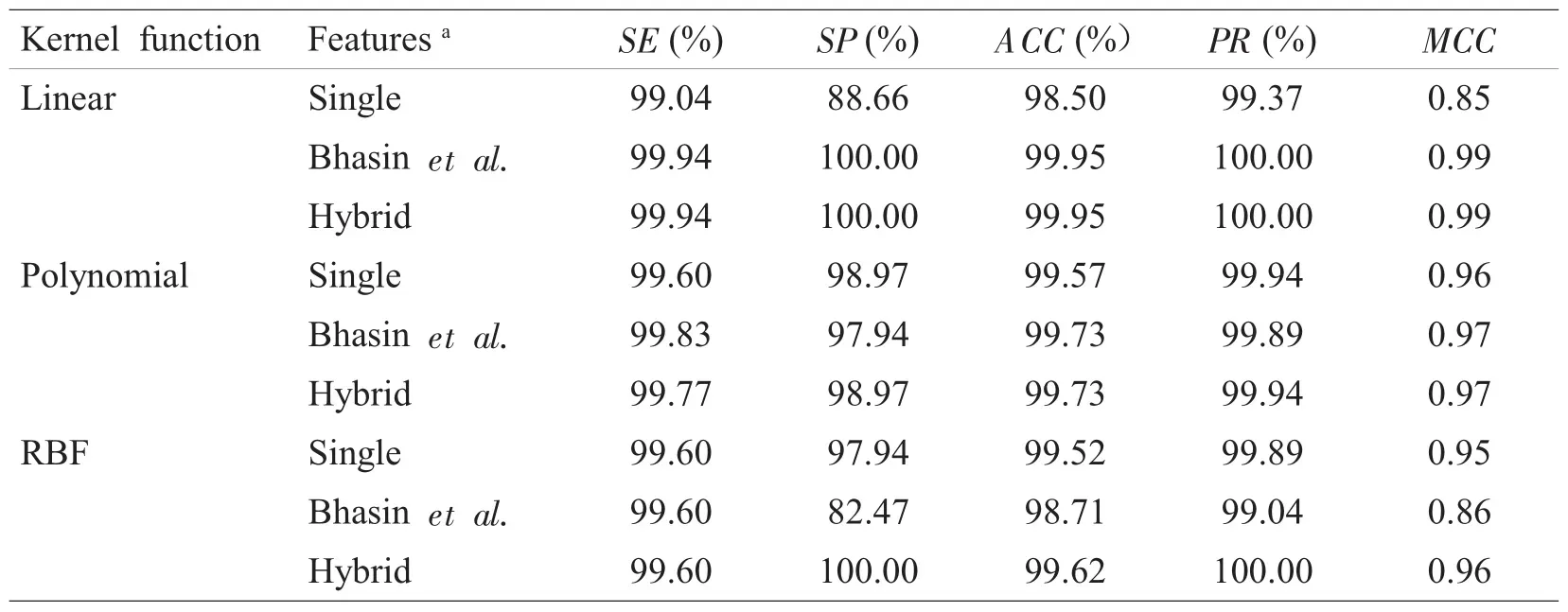
表1 GPCR蛋白序列的预测结果Table 1Performance of identifying GPCR sequences from decoys by SVM classifiers
2.2 GPCR分类预测
对于GPCR类型的预测,我们设计了5个分类器。当预测A类GPCR序列时,我们把A类数据作为机器学习的正类集,其余4类归为负类集,用同样的十倍交叉验证的方法,使用不同的序列特征和不同的SVM核函数,对数据进行训练学习,对于B、C、D、E类序列的识别也采用类似方法。所有实验结果如表2至表6所示,SVM模型参数设定与2.1节相同。
当预测A类GPCR序列时,除了使用单联氨基酸频率作为分类器特征时的预测效果不是很理想之外,采用Bhasin等的双联氨基酸使用频率和我们提出的Hybrid feature时,分类器都得到了很好的预测效果,预测准确率(ACC)都在99.60%以上(表2)。
从表3可以得知,当对B类GPCR序列进行预测时,使用单联氨基酸频率作为分类器特征,其预测效果也不是很好,而采用Bhasin等的双联氨基酸使用频率和我们提出的Hybrid feature构建的分类器,都得到了非常不错的预测效果,且总的来说,Hybrid feature构建的分类器要略优于Bhasin等的方法。
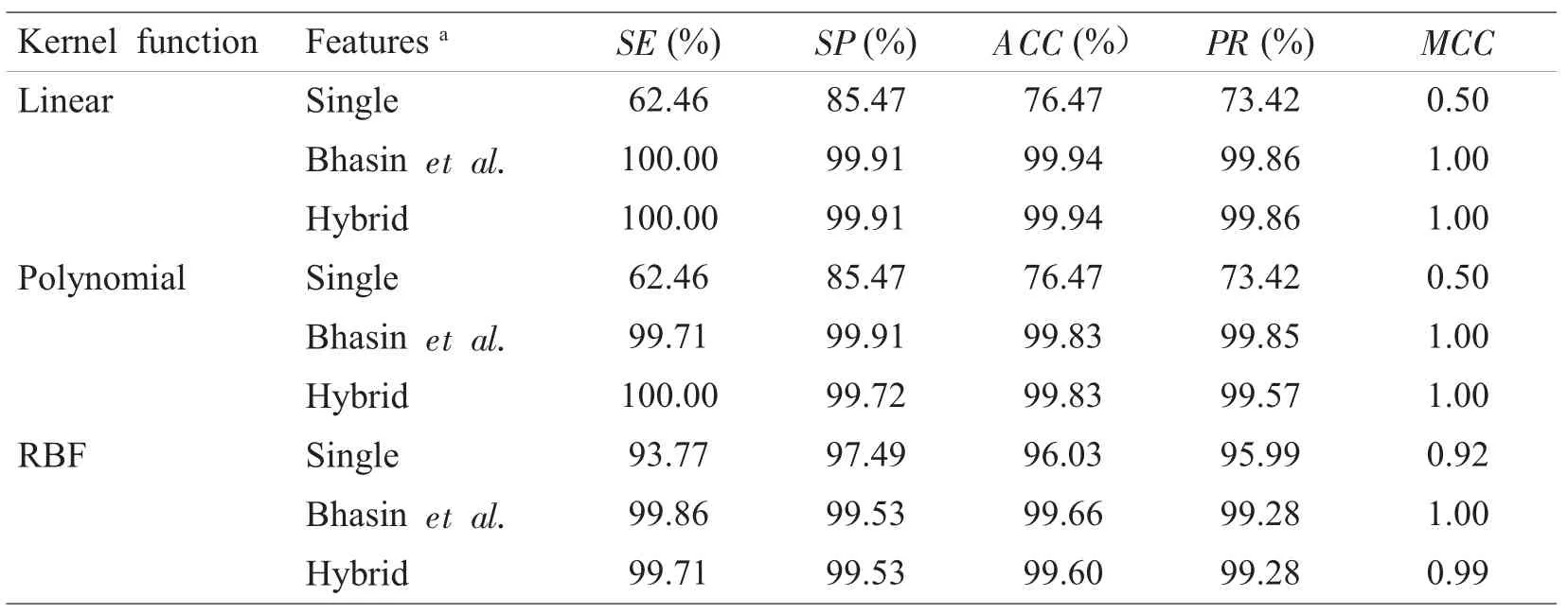
表2 A类GPCR蛋白序列的预测结果Table 2Performance of recognizing class A of GPCR sequences by SVM classifiers
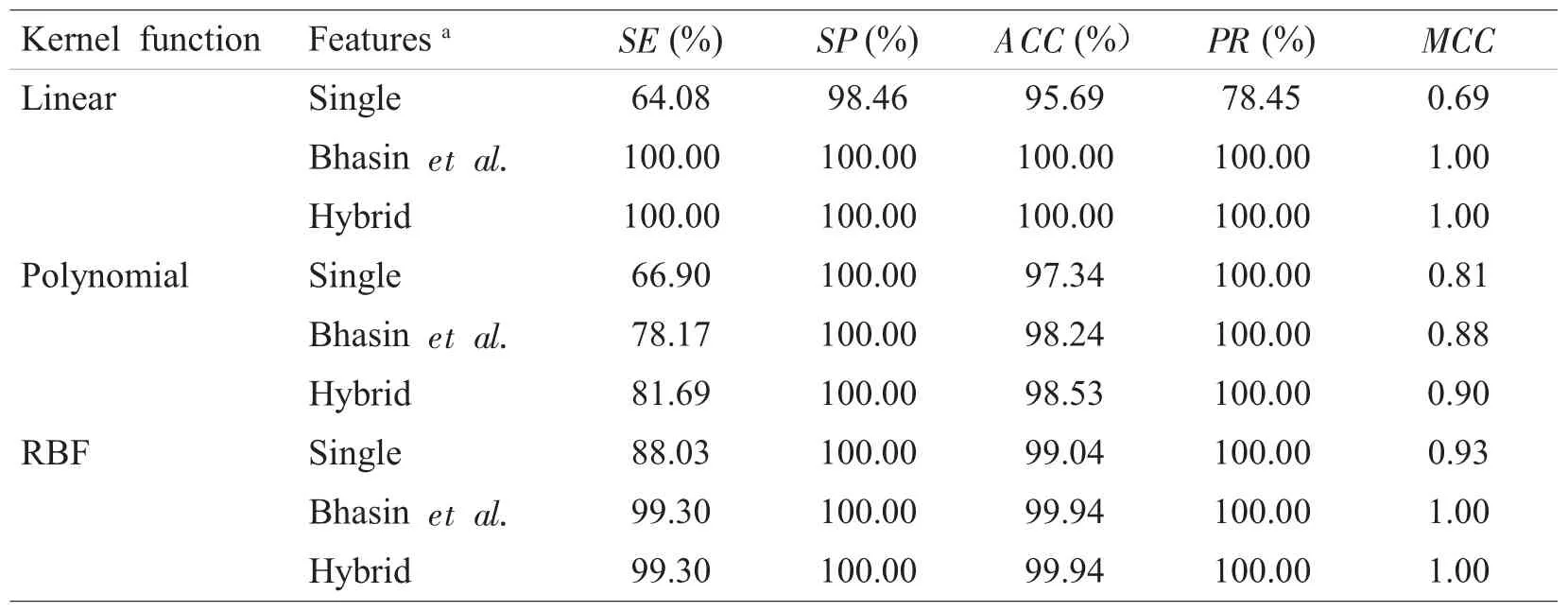
表3 B类GPCR蛋白序列的预测结果Table 3Performance of recognizing class B of GPCR sequences by SVM classifiers
当对C类GPCR序列进行预测时,用单联氨基酸频率构建的分类器,其预测效果也不是特别理想;而采用Bhasin等的双联氨基酸使用频率得到了最好的预测效果,预测准确率(ACC)均为99.89%。另外,我们也注意到,当使用我们提出的Hybrid feature并利用径向基核函数时,分类器也得到了很好的预测效果,预测准确率(ACC)为99.15%(表4)。
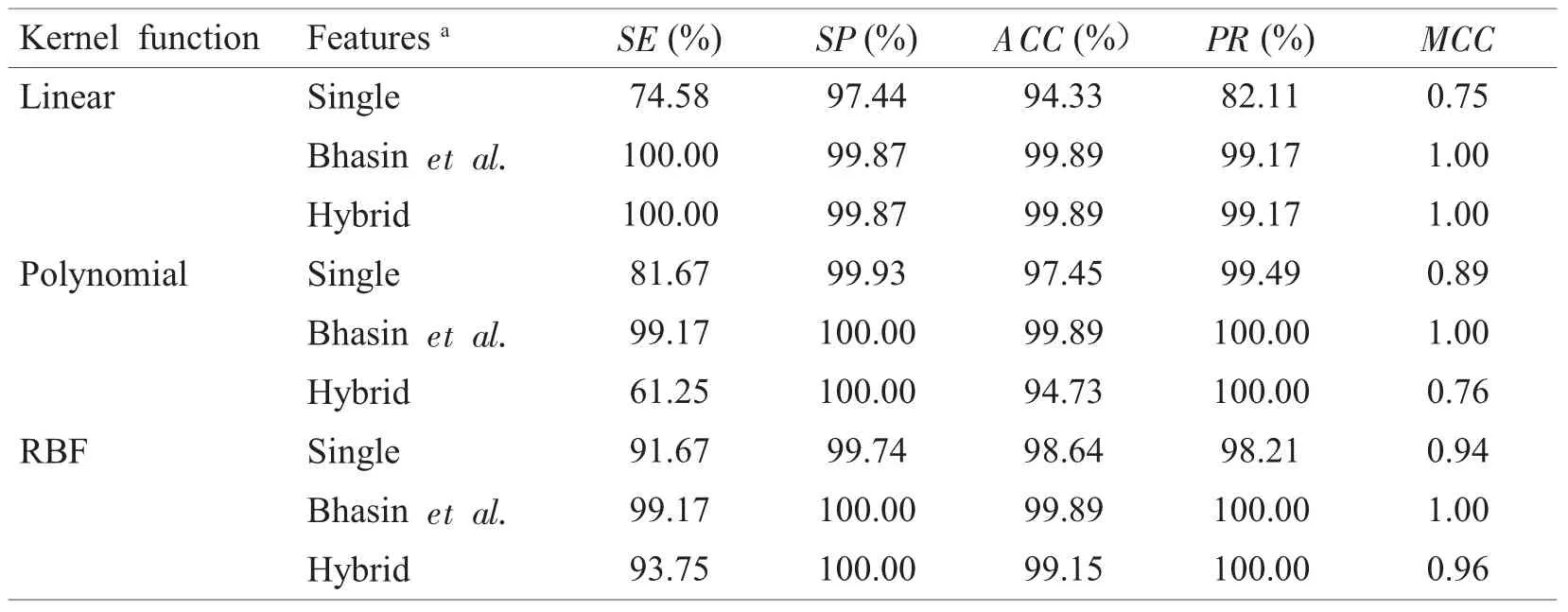
表4 C类GPCR蛋白序列的预测结果Table 4Performance of recognizing class C of GPCR sequences by SVM classifiers
表5中显示,当预测D类GPCR序列时,使用Bhasin等的双联氨基酸使用频率以及我们提出的Hybrid feature构建分类器,其预测效果均要优于单联氨基酸频率,且Hybrid feature的预测效果要略好于双联氨基酸使用频率。
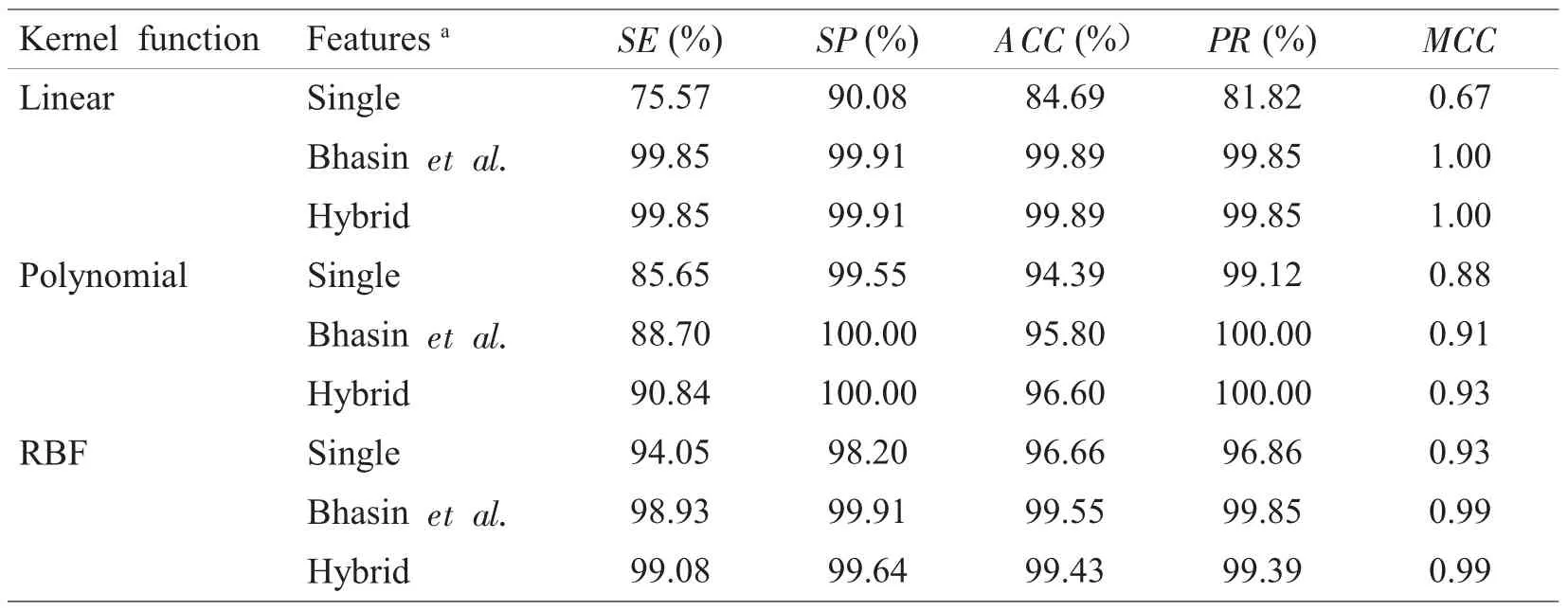
表5 D类GPCR蛋白序列的预测结果Table 5Performance of recognizing class D of GPCR sequences by SVM classifiers
当预测E类GPCR序列时,利用单联氨基酸频率作为分类器特征时预测效果不佳,而使用Bhasin等的双联氨基酸使用频率和我们的Hybrid feature时,分类器的预测效果有了明显的提高,且Hybrid feature的预测效果同样要略优于双联氨基酸使用频率(表6)。
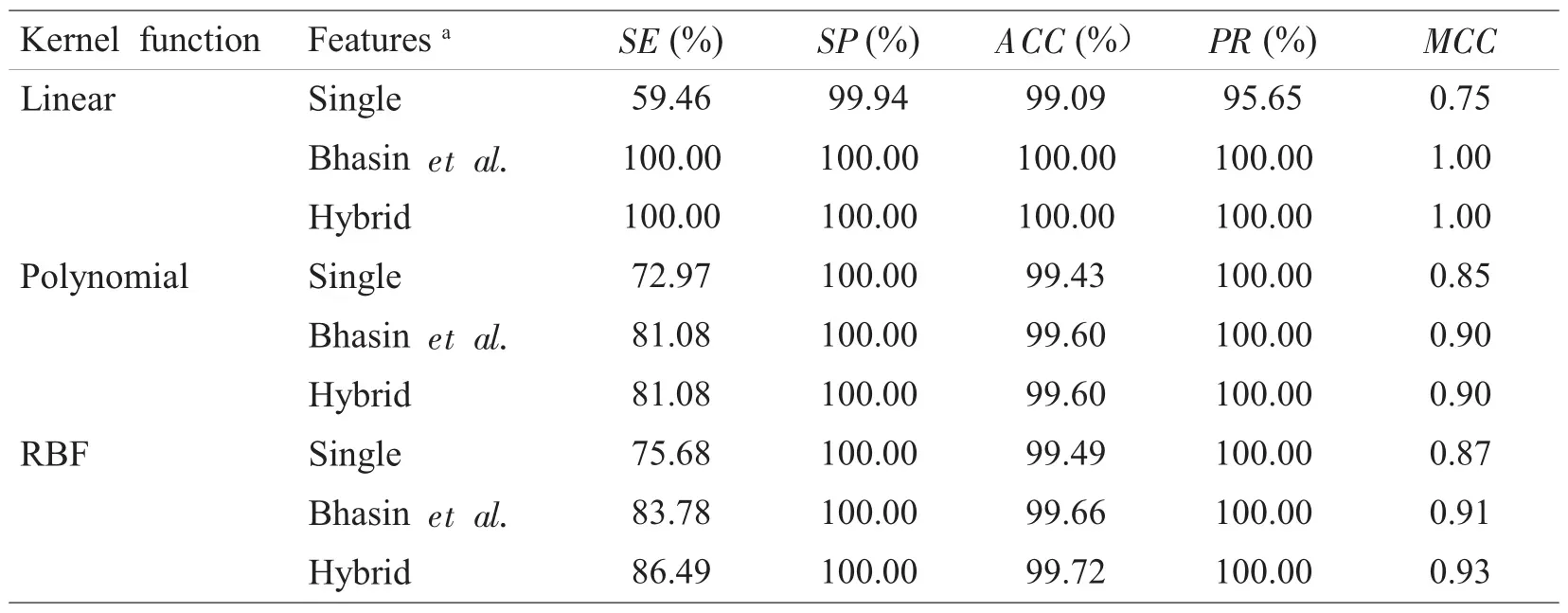
表6 E类GPCR蛋白序列的预测结果Table 6Performance of recognizing class E of GPCR sequences by SVM classifiers
从表2至表6可以看出,对各类GPCR序列进行预测时,当使用我们提出的Hybrid feature作为特征,且以径向基核作为核函数时,分类器都取得了非常不错的预测效果。因此,在设计基于SVM的GPCR类型分类器时,建议使用Hybrid feature为序列特征,同时采用径向基核核函数。但是,也应该看到,Bhasin等提出的这种基于SVM并提取双联氨基酸使用频率作为序列特征的分类器,也是一种很优秀的GPCR分类预测工具[7],它在GPCR类型的识别上也有着重要的实用意义。
3 讨论
我们知道,基因的绝对密码子使用频率与基因的功能类型有关[15~17]。在本文中,我们正是在这一研究结果的基础上,利用支持向量机对GPCR蛋白序列进行识别和分类的。事实上,利用单联或者双联氨基酸使用频率对GPCR的蛋白序列进行识别分类,也反映了氨基酸组成与蛋白功能类型的相关性,此前的很多研究报道已经表明,功能相似的蛋白质具有相似的氨基酸组成[15~17]。
为了进一步说明本文中用到的单联氨基酸使用频率、双联氨基酸使用频率和绝对密码子使用频率这3种序列特征与GPCR序列分类间的相关性,我们分别就3种特征作了主成分分析(图1),其中的图A、图B和图C分别对应于单联氨基酸使用频率、双联氨基酸使用频率以及绝对密码子使用频率,图中的A、B、C、D、E对应于5类GPCR序列,Decoy对应于99条欺骗序列。
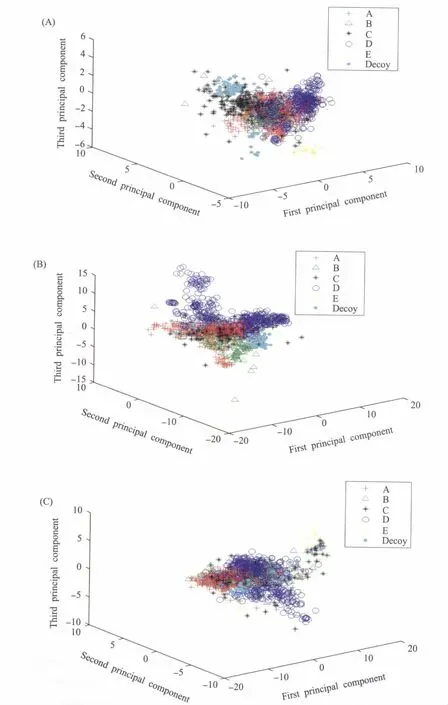
图1 五种类型的GPCR蛋白质序列及欺骗序列(decoy)的主成分分析图(A)单联氨基酸频率作为序列特征;(B)双联氨基酸作为序列特征;(C)绝对密码子使用频率作为序列特征Fig.1Dot plot of the three most dominant axes generated with PCA analysis method for five kinds of GPCR sequences and decoys(A)Single amino acid use frequency as features; (B)Double amino acid use frequency proposed by Bhasin et al.as features;(C)Hybrid feature combining codon use frequencies of mRNA genes and double amino acid use frequencies
从图1可以看出,99条欺骗序列在利用单联氨基酸使用频率(图1A)、双联氨基酸使用频率(图1B)所作的主成分分析图中聚集较为集中,与GPCR序列区分较为明显,因此,对于GPCR序列的识别问题,在设计SVM分类器时,提取各种特征和利用各种SVM核函数分类效果都十分理想(表1)。
对于GPCR序列分类的问题,从图1可以看出,就单联氨基酸使用频率、双联氨基酸使用频率,以及绝对密码子使用频率3种序列特征而言,5类GPCR序列之间显然均是非线性可分的关系。因此,我们采用支持向量机的方法,将非线性可分的样本提升到高维空间,对GPCR序列进行分类。从表2至表6可知,当联合使用Bhasin等的双联氨基酸使用频率及我们的Hybrid feature且利用线性核函数时,分类器都得到了非常好的预测效果,这和图1B和图1C的主成分分析结果有些矛盾,这表明本文在使用线性核函数构建分类器的过程中可能存在过度拟合的问题。从图1B可以看出,在利用双联氨基酸使用频率所作的主成分分析图中,A、B和D类GPCR序列聚集较为集中,区分较为明显,所以使用双联氨基酸使用频率为特征构建分类器时,分类效果较好,而E类GPCR序列与其它GPCR序列区分不明显,在分类预测时效果不佳。但当我们使用绝对密码子使用频率进行主成分分析时,E类GPCR序列聚集集中,与其它的GPCR序列区分明显,所以在分类预测时,加入绝对密码子使用频率的信息,预测效果得到了提高(表6)。因此,综合考虑所有类型分类器的识别效果,在GPCR序列类型预测的问题中,设计SVM分类器时,最佳方案是选择包含基因序列绝对密码子使用频率和蛋白序列双联氨基酸使用频率两部分信息的组合特征(Hybrid feature)作为SVM的输入,同时使用径向基核作为核函数。
致谢:感谢东南大学生物电子学国家重点实验室的孙啸教授对本工作的指导。
1.Bockaert J,Pin JP.Molecular tinkering ofGproteincoupled receptors:anevolutionary success.EMBOJ, 1999,18(7):1723~1729
2.Horn F,Mokrane M,Weare J,Vrien G.G-protein coupled receptors or the power of data.Genomics and proteomics: functional and computational aspects.New York:Kluwer Academic/Plenum,2000,192~214
3.Lapinsh M,Gutcaits A,Prusis P,Post C,Lundstedt T, Wikberg JE.Classification of G-protein-coupled receptors by alignment independent extraction of principal chemical properties of primary amino acid sequences.Protein Sci, 2002,11(4):795~805
4.Joost P,Methner A.Phylogenetic analysis of 277 human G-protein-coupled receptors as a tool for the prediction of orphan receptor ligands.Genome Biol,2002,3(11):1~16
5.候永丰,李通化.HMM用于G蛋白偶联受体超家族的识别.同济大学学报(自然科学版),2004,32(12):1696~1700 Hou YF,Li TH.Classifying G-protein coupled receptors with hidden Markov models.Journal ofTongji University (Natural Science),2004,32(12):1696~1700
6.Karchin R,Karplus K,Haussler D.Classifying G-protein coupledreceptorswithsupportvectormachines. Bioinformatics,2002,18(1):147~159
7.BhasinM,RaghavaGP.GPCRpred:anSVM-based methodforpredictionoffamiliesand subfamilies of G-protein coupled receptors.Nucleic Acids Res,2004,32: W383~W389
8.Zhou T,Weng JH,Sun X,Lu ZH.Support vector machine for classification of recombination hotspots and coldspots in Saccharomyces cerevisiaebased on codon composition. BMC Bioinformatics,2006,7:223
9.Wu JS,Hu MJ,Zhou T,Weng JH,Jiang P,Sun X. Support vector machine for prediction of siRNA silencing efficacy.Journal of Southeast University(English Edition),2006,22(4):501~504
10.吴建盛,谢建明,周童,翁建洪,孙啸.基于支持向量机的细菌基因组水平转移基因预测.生物物理与生物化学进展,2007, 34(7):724~731 Wu JS,Xie JM,Zhou T,Weng JH,Sun X.Support vector machineforpredictionofhorizontalgenetransfersin bacteria genomes.Prog Biochem Biophys,2007,34(7): 724~731
11.Horn F,Weare J,Beukers MW,Hrsch S,Bairoch A, Chen W,Edvardsen O,Campagne F,Vriend G.GPCRDB: aninformationsystemforGprotein-coupledreceptors. Nucleic Acids Res,1998,26(1):275~279
12.Vapnik V.The nature of statistical learning theory.New York:Springer-Verlag,1995.1~188
13.Dimitriadou E,Hornik K,Leisch F,Meyer D,Weingessel A. e1071:Miscfunctionsofthedepartmentofstatistics (e1071).TU Wien,R package,Version 1.5-16.Available from http://cran.R-project.org,2007
14.Matthews BW.Comparison of the predicted and observed secondarystructureofT4phagelysozyme.Biochim Biophys Acta,1975,405(2):442~451
15.Ma JM,Zhou T,Gu WJ,Sun X,Lu ZH.Cluster analysis of the codon use frequency of MHC genes from different species.Biosystems,2002,65(2-3):199~207
16.Zhou T,Gu WJ,Ma JM,Sun X,Lu ZH.Analysis of synonymouscodonusageinH5N1virusandother influenza A viruses.Biosystems,2005,81(1):77~86
17.Gu WJ,Zhou T,Ma JM,Sun X,Lu ZH.Analysis of synonymous codon usage in SARSCoronavirusand other virusesintheNidovirales.VirusRes,2004,101(2): 155~161
Abstract:G-protein coupled receptor is a very important signal molecule receptor and its dysfunction may lead to the emergence of many diseases.According to the previous studies,a method combining the feature analysis methods of sequences with support vector machine(SVM)technology was proposed for identifying GPCRs and their type by analyzing the characteristics of sequence differences.Especially,codon use frequencies of mRNA genes translating into GPCR proteins were first selected as the sequence feature,in respect that it is the inherently the fusion of both codon usage bias and amino acid composition signals. The results showed that the optimal SVM classifiers for predicting GPCR sequences and their type were designed by choosing the hybrid feature by combining codon use frequencies of mRNA genes and double amino acid use frequencies and using the RBF kernel as kernel function after considering the performance of all types of SVM classifiers.
Key Words:G-protein coupled receptor(GPCR);Support vector machine(SVM);Codon use frequency (FCU)
Prediction of G-Protein Coupled Receptors and Their Type
WU Jiansheng1,MA Xin2,ZHOU Tong2,TANG Lihua1,HU Dong1
1.School of Geography and Biological Information,Nanjing University of Posts and Telecommunications, Nanjing210046,China;
2.State Key Laboratory of Bioelectronics,Southeast University,Nanjing 210096,China
Q516
2009-11-03;接受日期:2010-01-24
南京邮电大学科研启动基金项目(NY209027),南京邮电大学青蓝计划项目(NY206060)
胡栋,电话:(025)85885169,E-mail:hud@njupt.edu.cn
This work was supported by grants from Research Start-up Funding by Nanjing University of Posts and Telecommunications (NY209027)and“QingLan”Project of Nanjing University of Posts and Telecommunications(NY206060)
Received:Nov 3,2009Accepted:Jan 24,2010
Corresponding author:HU Dong,Tel:+86(25)85885169,E-mail:hud@njupt.edu.cn
猜你喜欢
广西植物(2022年8期)2022-09-07
电子产品世界(2022年4期)2022-04-21
汽车观察(2021年4期)2021-05-10
计算机系统应用(2021年2期)2021-02-23
西部交通科技(2021年9期)2021-01-11
发明与创新·中学生(2019年6期)2019-06-26
计算机测量与控制(2019年4期)2019-05-08
新西部下半月(2018年6期)2018-09-26
生物学教学(2018年2期)2018-08-07
安徽农业科学(2018年1期)2018-05-14
