
潜在的星际分子PSiCP势能面的密度泛函理论研究
2010-09-01 03:02:54张凤华刘慧玲
石油化工高等学校学报 2010年1期
李 飞, 王 婵, 张凤华, 刘慧玲
(1.辽宁石油化工大学化学与材料科学学院,辽宁抚顺113001; 2.吉林大学理论化学研究所理论化学计算国家重点实验室,吉林长春130021)
潜在的星际分子PSiCP势能面的密度泛函理论研究
李 飞1, 王 婵1, 张凤华1, 刘慧玲2
(1.辽宁石油化工大学化学与材料科学学院,辽宁抚顺113001; 2.吉林大学理论化学研究所理论化学计算国家重点实验室,吉林长春130021)
潜在的单重态星际分子PSiCP的势能面在密度泛函和从头算理论水平下进行计算。在B3LYP/6-311G(d)水平下,共得到8个异构体,它们由10个过渡态所连接。在QCISD/6-311G(d)水平下,3个异构体cSiPCP 1,PSiCP 7和SiCPP 8具有较大的转变能垒,是动力学稳定的异构体。分析得到的3个异构体的结构性质和光谱学参数可为星际探测和制备这些异构体提供理论依据。
势能面; PSiCP; 稳定性; 密度泛函; 理论研究
硅、碳和磷化学近年来在天体物理和微电子材料制备工艺的研究领域内引起了诸多的重视。到目前为止,许多含有Si和P元素的分子或自由基,如SiCn(n=1-4)、SiP、PO和CP等在星际空间中已经被探测到[1-2]。这些分子的发现是的化学家认为在星际分子云中可能存在更加复杂的含有C、Si和P元素的分子或自由基。另外,含有Si和C元素的相关材料还引起了来自于微电子材料制备领域内的重视。众所周知,二元的SiC化合物经常被用作半导体材料应用于微电子和光电子的器件中[3]。磷元素可被用作少量的掺杂剂来改变硅碳半导体材料的某些特性。大量的实验和理论计算已经对SimCn和SimPn等一些小分子进行了系统的研究[4-10]。本文将研究单重态 PSiCP分子体系。通过理论计算建立它的势能面来预测PSiCP异构体的稳定性。另外,详细的理论计算还可以预测PSiCP分子的光谱学特性和结构性质。这些将有利于 PSiCP分子的未来星际探测和实验室制备。
1 计算方法
所有的理论计算都是使用 Gaussian 98程序包完成的。首先在B3LYP/6-311G(d)水平下得到异构体和过渡态的几何结构,再在CCSD(T)/6-311G(2d)水平下对其进行的单点能量的计算。在B3LYP/6-311G(d)水平下,对过渡态进行内禀反应坐标(IRC)计算确认连接正确的异构体。对于重要异构体在QCISD/6-311G(d)水平下进一步优化计算,然后在CCSD(T)/6-311+G(2df)水平下计算其单点能。相对能量的计算包含了零点振动能校正。
2 PSiCP的势能面
2.1 PSiCP的势能面
在B3LYP/6-311G(d)水平下,计算得到的8个单重态异构体和过渡态的几何构型见图1。由图1可见,它们分别是直线型异构体 PSiCP 7[1∑](79.9,99.5)和SiCPP 8[1∑](163.4,171.8)、4个四元环状的异构体cSiPCP 1[1A1](0.0,0.0), cPSiCP 2(115.4),cSiPCP 3[1A′](137.9), cPPCSi 4(164.3)和2个三元环状异构体的 SicCPP 5[1A1](9 9.0),Si-cPPC 6[1A′](245.4)。需要说明的是方括号内的符号代表着异构体的电子态,括号内的第1个和第2个数值分别代表着异构体,在 CCSD(T)//B3LYP和 CCSD (T)//QCISD水平下,计算得到相对于异构体1 (0.0,0.0)的相对能量,单位是kJ/mol。

Fig.1 Optimized geometries of PSiCP isomers and transition states at the B3LYP/6-311G(d)level图1 在B3LYP/6-311G(d)水平下优化PSiCP异构体和过渡态的几何构型
一般来说,一个异构体的最低的异构化或解离转变能垒的大小决定着该异构体的动力学稳定性,具有较大的转变能垒的异构体具有较高的动力学稳定性。图2为 在CCSD(T)//B3LYP水平下计算得到单重态的PSiCP异构体的势能面示意图。由图2可以看出,对于 PSiCP异构体来说,从势能面上看,没有异构体可以解离成相应的碎片的路径。因此,异构化转变过程决定着这些PSiCP异构体的动力学稳定性。从势能面上的相对能量和转变能垒上看,3个异构体cSiPCP 1,PSiCP 7和SiCPP 8由于具有相对较大的异构化能垒(大于70 kJ/mol)。3个异构体cSiPCP 1、PSiCP 7和SiCPP 8分别具有126.6(127.1)(1→TS1/2→2)、73.9(75.2)(7→TS1/7→1)和117.9(120.8)(8→TS5/8→5)kJ/ mol的转变能垒。这样大的转变能垒足以保证异构体1、7和8存在于实验室和星际空间中的低温环境中(例如致密星云中)。而且,在CCSD(T)//QCISD水平下,计算得到的异构体1、7和8的转变能垒(括号内的数值)与CCSD(T)//B3LYP水平下的计算值在3.0 kJ/mol的误差内。这说明两种理论水平下计算的结果都是非常可靠的。然而,其它剩下的异构体在研究中就不是主要的研究对象。这主要是由于它们具有较小的转变能垒(小于40 kJ/mol),这么小的转变能垒不能足以保证这些异构体稳定地存在于PSiCP异构体的势能面上,它们能够通过各自的异构化路径很容易地转变成稳定的异构体。

Fig.2 Schematic potential energy surface of PSiCP at the CCSD(T)//B3LYP level图2 在CCSD(T)//B3LYP水平下计算得到单重态的PSiCP异构体的势能面示意图
2.2 稳定异构体的结构性质
相对能量最低的异构体cSiPCP 1具有C2v的对称性和1A1的电子态。它的轨道对称性为{core}其中1b1是π轨道,主要离域在整个分子中PCP的3个原子上的。而1a2是π反键轨道。计算得到的SiP(0.232 4 nm)键长稍微长于标准的Si—P单键键长(0.228 2 nm, H3Si—PH2)。SiC(0.195 2 nm)键长也长于标准的Si—C单键键长(0.188 5 nm,H3Si—CH3)。外围的两个CP(0.167 6 nm)键长非常接近于标准的CP双键(0.167 nm,H2CPH)。从键长和NBO分析看,异构体1应该是式(1)、(2)中的几种共振结构形式。其中结构(1)是其存在的主要形式。符号“—”代表着孤对电子。
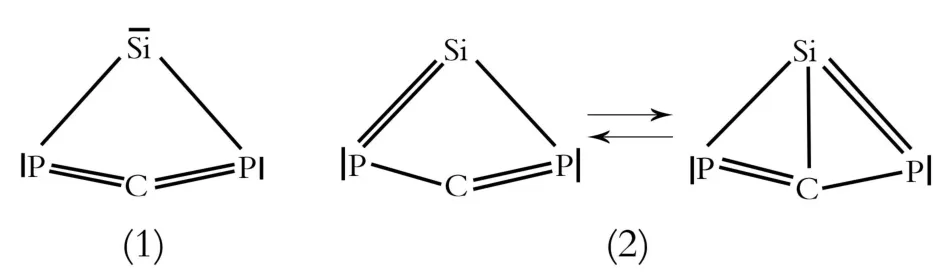
直线型异构体PSiCP 7具有1∑电子态。它的PSi键长(0.196 9 nm)和中间的CP键长(0.155 8 nm)非常接近于各自相应的 Si≡P三键(0.195 7 nm,HSi≡P)和C≡P三键(0.153 9 nm,HC≡P)的键长。中间的SiC键长(0.177 4 nm)介于SiC双键(0.1 7 07 nm,H2CSiH2)和Si—C单键(0.188 5 nm,H3C—SiH3)键长之间。它的轨道对称性可以表示为{core}1σ22σ23σ24σ21π45σ22π4。1π轨道主要离域在SiCP三个原子上。而2π轨道主要定域在CP和SiP键上。依据键长和NBO分析的结果,直线型异构体7是下面的结构形式。
从对异构体7的价键结构看,它是含有Si≡P和C≡P结构的异构体。直到目前为止,在实验室的材料制备中还没有合成出含有Si≡P和C≡P三键结构的化合物。异构体7的计算结果将为今后的实验制备含有Si≡P或C≡P三键结构的化合物提供理论依据。

直线型异构体 SiCPP 8也具有1∑的电子态。它的SiC键长(0.168 4 nm)介于标准的Si=C双键(0.170 7 nm,H2C=SiH2)和Si≡C三键(0.164 7 nm,HSi≡CH)键长之间。中间CP键长(0.162 0 nm)介于CP双键(0.167 0 nm,H2CPH)和C≡P三键(0.153 9 nm,HC≡P)之间。末端的PP键长(0.191 5 nm)稍微长于 P≡P三键(0.189 8 nm,P≡P)键长。它的轨道对称性也是{core} 1σ22σ23σ24σ21π45σ22π4。1π轨道主要离域在整个SiCPP链上,稍微偏重于内部的CP键上。2π轨道主要定域在SiC和PP键上。依据键长和NBO的分析,直线型异构体8应该是下面的结构形式。

最后,期望计算得到的稳定异构体的光谱学参数,即振动频率、偶极距和转动常数见表 1。在QCISD/6-311G(d)水平下,3个异构体1,7和8的主振动频率分别是1 136、1 480和1 439 cm-1。它们对应的振动强度分别是242、18、897 km/mol。异构体1和7分别具有较大的偶极距1.548 2 D和 3.106 1 D。这些光谱学参数对未来PSiCP异构体的光谱识别具有指导意义。

表1 在QCISD/6-311G(d)水平下计算得到的PSiCP的稳定异构体的光谱学参数Table 1 Spectroscopic parameters the kinetically stable PSiCP isomers at the QCISD/6-311G(d)level
[1]McCarthy M C,Thaddeus P.Microwave and laser spectroscopy of carbon chains and rings[J].Chem.soc.rev.,2001, 30(3):177-185.
[2]Kaiser R I.Experimental investigation on the formation of carbon-bearing molecules in the interstellar medium via neutral-neutral reactions[J].Chem.rev.,2002,102(5):1309-1358.
[3]Furthmüller J,Bechstedt F,Hüsken H,et al.Si-rich SiC(111)/(0001)3×3 and sqrt[3]×sqrt[3]surfaces:A motthubbard picture[J].Phys.rev.B,1998,58(20):13712-13716.
[4]Rittby C M L.An ab initio study of the structure and infrared spectrum of Si3C[J].J.chem.phys.,1992,96(9):6768 -6772.
[5]Karni M,Apeloig Y,Schröder D,et al.HCSiF and HCSiCl:The first detection of molecules with formal C≡Si triple bonds[J].Angew.chem.int.ed.1999,38(3):331-335.
[6]Bertolus M,Finocchi F,MilliéP.Investigating bonding in small silicon–carbon clusters:Exploration of the potential energy surfaces of Si3C4,Si4C3,and Si4C4using ab initio molecular dynamics[J].J.chem.phys.,2004,120(9):4333 -4343.
[7]Nam S H,Park S M.Interaction of ultraviolet laser with a silicon carbide plume produced by laser ablation[J].J.appl.phys.,2004,95(12):8425-8430.
[8]Jakubek ZJ,Nakhate S G,Simard B.The SiP molecule:The first observation and spectroscopic characterization[J].J.chem.phys.,2002,116(15):6513-6520.
[9]Chong D P.Local density studies of diatomic AB molecules,A,B=C,N,O,F,Si,P,S,and Cl[J].Chem.phys.lett.,1994,220(1-2):102-108.
[10]张凤华,王婵,李飞,等.[Si,O,C,P]自由基的结构和生成路径[J].辽宁石油化工大学学报,2009,29(4):7-11.
(Ed.:YYL,Z)
Theoretical Study on Potential Energy Surface of the Singlet Promising Interstellar Molecule PSiCP
LI Fei1,WANG Chan1,ZHANG Feng-hua1,LIU Hui-ling2
(1.School of Chemistry&Materials Science,Liaoning Shihua University,Fushun Liaoning113001,P.R.China; 2.State Key Laboratory ofTheoretical and Computational Chemistry,Institute ofTheoretical Chemistry, Jilin University,Changchun Jilin130023,P.R.China)
17April2009;revised28October2009;accepted11November2009
The potential energy surface of the promising interstellar molecule PSiCP was calculated at the density functional theory and ab initio level.At the B3LYP/6-311G(d)level,eight isomers connected by ten transition states were located on the potential energy surface.At the QCISD level,three isomers cSiPCP 1,PSiCP 7 and SiCPP 8 possess considerable kinetic stability.The bonding nature and spectroscopic parameters were analyzed,which will be helpful for the future astrophysical detection and the synthesis in the laboratory.
Potential energy surface;PSiCP;Stability;Density functional theory;Theoretical study
.Tel.:+86-413-6860548;fax:+86-413-6860548;e-mail:lnpulf@126.com
O643
A
10.3696/j.issn.1006-396X.2010.01.011
2009-04-17
李飞(1981-),男,辽宁沈阳市,讲师,博士。
辽宁省科技厅资助项目(20091048)。
1006-396X(2010)01-0043-4
猜你喜欢
燃料化学学报(2023年3期)2023-03-11 03:34:40
系统仿真技术(2022年4期)2023-01-17 13:01:44
云南化工(2021年8期)2021-12-21 06:37:38
中学课程辅导·教学研究(2021年8期)2021-07-14 13:44:52
燃料化学学报(2021年5期)2021-06-02 14:01:38
青岛大学学报(工程技术版)(2019年2期)2019-09-10 07:22:44
国外医药(抗生素分册)(2016年4期)2016-07-12 14:25:19
信息记录材料(2016年4期)2016-03-11 15:22:30
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
