
原子转移自由基活性可控聚合进展
2010-08-28 06:38方旭东周博尤伟张蕊李贤宇
天津化工 2010年4期
方旭东,周博,尤伟,张蕊,李贤宇
(1.天津渤海职业技术学院 天津 300402;2.汉维艾施格科技(北京)有限公司 北京 100101)
原子转移自由基活性可控聚合进展
方旭东1,周博1,尤伟2,张蕊1,李贤宇1
(1.天津渤海职业技术学院 天津 300402;2.汉维艾施格科技(北京)有限公司 北京 100101)
活性自由基聚合是目前高分子科学中最为活跃的研究领域之一,原子转移自由基聚合(ATRP)反应是实现活性聚合的一种颇为有效的途径,也是高分子化学领域的最新研究进展之一。介绍了ATRP反应的特点、聚合反应机理、应用及前景展望。
原子转移;自由基;聚合;应用;前景
自由基聚合有很多优点,适合自由基聚合的单体多,大量应用的高分子材料大部分通过自由基聚合得到,但由于自由基聚合存在链转移和链终止反应,传统自由基聚合不能较好地控制相对分子质量、相对分子质量分布及大分子结构,从而影响高分子材料的力学、电学等性能,限制了高分子材料的使用范围。而活性聚合具有无终止无转移、引发速率远大于链增长速率等特点,与传统自由基聚合相比能更好地实现对分子结构的控制,是实现分子设计、合成具有特定结构和性能聚合物的重要手段。
1995年,王锦山博士[1]首次提出了原子转移自由基聚合,由于这种自由基聚合反应具有聚合过程活性可控,能够合成低分散度和确定相对分子质量及分子结构的聚合物,引起了世界各国高分子学家的极大兴趣,纷纷开展该领域研究,取得了许多创新性的研究成果,显示了良好的发展前景。
1 原子转移自由基聚合(ATRP)的特点
20世纪50年代配位聚合技术的出现,开辟了立构规整聚合的新纪元;而各种活性聚合技术的发展为合成出结构和组成可控的聚合物材料提供了技术基础。自由基聚合产品占了所有聚合物产品的一半以上,因此,发展“可控、活性自由基聚合”成为人们梦寐以求的目标。自1995年中国旅美学者王锦山等首先发明原子转移自由基聚合(ATRP)技术后,立即引起世界各国高分子界专家学者和工业界的极大兴趣。原子转移自由基聚合技术是近几年迅速发展并有着重要应用价值的一种活性聚合技术,可有效地对聚合物的分子结构进行设计,制备出各种不同性能、不同功能的新型聚合物材料,即所谓的“量体裁衣”[2]。它可以通过分子设计制得多种具有不同拓扑结构(线型、梳状、网状、星形、树枝状大分子等)、不同组成和不同功能的结构确定的聚合物及有机无机杂化材料。与离子聚合等传统活性聚合技术相比,它具有单体覆盖面广,聚合条件温和,易于实现工业化生产等显著优点,将成为合成新型高分子材料的一个新方向。其产品在高性能粘合剂、分散剂、表面活性剂、高分子合金增溶剂和加工助剂、热塑性弹性体、绿色化学品、电子信息材料及新型含氟材料等高技术领域都具有广泛的应用前景[3]。从20世纪90年代开始,高分子化学家着重于研究通过化学方法实现对自由基聚合的控制,这些方法具有广泛的适用性。ATRP的独特之处在于使用了卤代烷作引发剂,并用过渡金属催化剂或退化转移的方式,有效地抑制了双基终止反应。由于动力学原因,在自由基聚合中完全消除终止反应是不可能的。准确地说,原子转移自由基聚合方法应称为活性或受控自由基聚合。虽然不同活性自由基聚合采用的引发体系不同,但基本特征都是由活性种与某种媒介物可逆反应生成比较稳定的休眠种。两者之间存在动态平衡,此平衡必须大大倾向于休眠种一端,使自由基平衡浓度很低,大大抑制了双基终止反应。活性种和休眠种之间相互转变速率和增长速率之比是控制相对分子质量分布的重要因素,这一比值越高,相对分子质量分布越窄[4]。与传统的活性聚合如阴(或阳)离子聚合和基团转移聚合(GTP)相比,ATRP可以同时适用于非极性和极性单体,如苯乙烯(St)、二烯烃类和(甲基)丙烯酸酯类单体,可以制备包括无官能团的均聚物及无规、嵌段、星形和梯度共聚物与超支化物(hyperbrench)、树枝状物(dentrimer)在内的诸多结构清晰的高分子化合物,其相对分子质量可以控制在103~105、MW/MN在1.05~1.5。
2 原子转移自由基聚合机理
ATRP聚合反应以过渡金属作为催化剂,使卤原子实现可逆转移,包括卤原子从烷基卤化物到过渡金属络合物(盐),再从过渡金属络合物(盐)转移至自由基的反复循环的原子转移过程,伴随着自由基活性(增长链自由基)种和大分子有机卤化物休眠种之间的可逆转换平衡反应,抑制着自由基活性种在较低的浓度,减少了增长链自由基之间的不可逆双分子终止副反应,使聚合反应得到有效的控制。ATRP的核心是引发剂卤代烷(RX)与单体中C—C链加成,加成物中C—X键断裂产生自由基,引发聚合。北京大学的袁金颖[2]等对不同引发剂结构对原子转移自由基聚合反应的影响进行了研究。
原子转移“活性”自由基聚合(ATRP)示意图如图:
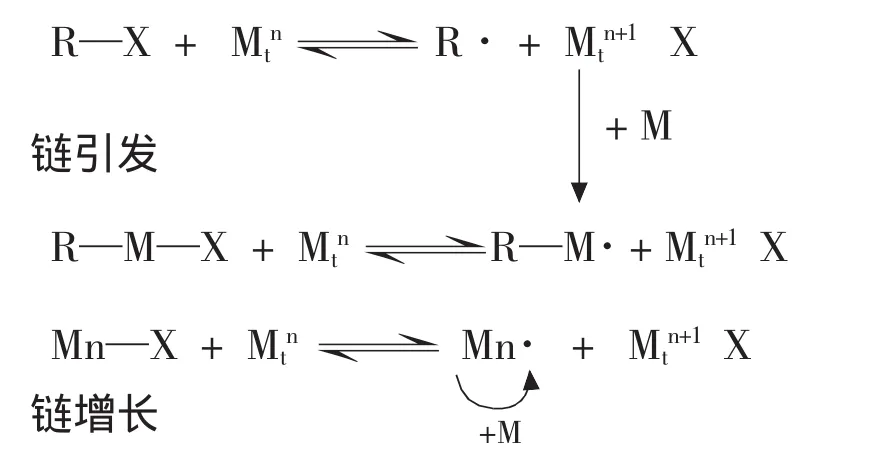
图1 原子转移“活性”自由基聚合(ATRP)反应
因为烷基卤化物(RX)对人体有较大的毒害,低氧化态过渡金属复合物易被空气中氧气氧化、储存较困难、价格高、不易得、不易处理等缺点,又发展了反向原子转移自由基聚合(reverse ATRP)。ATRP与反向ATRP在于引发剂类型不同,过渡金属卤化物氧化态不同。在反向ATRP聚合体系中,普通的自由基引发剂和高氧化态过渡金属复合物替代了ATRP聚合体系中使用的RX和低氧化态过渡金属复合物,聚合反应也是通过可逆的卤原子的原子转移反应得以控制。
ATRP聚合体系可制得无规共聚物、均聚物、嵌段共聚物、接枝共聚物、梯形、交替共聚物和星型聚合物。ATRP是实现“活性”控制自由基聚合的有效手段之一,其优势为:聚合迅速,聚合产物本身为ω2Cl原子的大分子引发剂,可在ATRP催化体系中进行共聚、均聚,在80~130℃范围内为合成复杂结构的聚合物提供较简便的方法,往往一步即可;催化剂可以简便分离,催化剂的数量仅影响反应速率,对相对分子质量并没有影响,所以可以使用较多催化剂来加速聚合反应。不过,ATRP催化剂的配体用量非常大,催化体系昂贵,聚合速度慢,有些单体如醋酸乙烯酯不能很好聚合,无法得到立构规整聚合物。目前研究开发新型经济实用的催化引发体系依然是ATRP的反应机理与应用研究的重点,对催化剂配体研究可以从根本上改善ATRP,并对催化剂数量的需求下降。ATRP适用单体范围之广,反应条件之温和,分子设计能力之强是现有的其它活性聚合无法比拟的。
3 ATRP技术的应用
3.1 制备分布较窄的均聚物
采用这类ATRP方法可以使苯乙烯、丙烯酸酯类、甲基丙烯酸酯类等单体聚合,合成相对分子质量103~105,且分布较窄的均聚物(Mw/Mn=1.04~1.55)。丙烯腈聚合时,由于高分子链上的腈基与铜催化剂配位,使反应速率逐渐降低,其动力学现象与其它单体有明显的差别。
3.2 制备具有特殊链端的聚合物
选择合适的单官能团或双官能团引发剂,可制备链端具有一两个卤原子的聚合物。卤原子可通过亲核取代反应转变成其它官能团,如—COOH、—OH、—NH2等。采用带有官能团的卤代物作引发剂,也可以在产物中引入多种官能团,如—COO H、—OH、—CN可聚合的乙烯基及作为标记的萘基和蒽基等。
3.3 在制备共聚物上的应用
利用ATRP制备嵌段共聚物的例子不甚枚举。如采用一锅法制备PS-PMMA[5]、聚甲基丙烯酸氧化乙烯酯-b-聚甲基丙烯酸丁酯;应用ATRP合成非烯烃类的单体[6](1-双环丁烷甲酸甲酯);以聚环氧乙烷为大分子引发剂,聚合甲基丙烯酸羟乙酯。制备聚苯乙烯-丙烯腈共聚物,并对其进行了扩链反应,制得了相对分子质量可控的含有丙烯腈的共聚物[7];制备具有相分离结构的PMMA-b-PBMA共聚物PS-PAA两亲性嵌段共聚物等。采用其它活性可控聚合与ATRP相结合也是制备共聚物的常用方法。如利用阴离子聚合MMA后,经Br2处理,再与MMA,St,BA制得双嵌段,三嵌段共聚物[8];在PS及PMS主链上引入氯甲基,溴甲基或2-溴丙酰基后,再接枝[9],以及用阴离子聚合与ATRP相结合,制备聚二甲基硅氧烷和聚(甲基)丙烯酸酯的AB、ABA型共聚物等。
在材料表面接技共聚,如在C60表面接枝PS[10];在Au基片表面接上引发剂后在室温下进行ATRP反应[8];在SiO2基片上经Cl3SiOCH2CH2phCH2Cl(p)处理,再与丙烯酰胺进行ATRP反应;在PS-b-PE-c-PB-b-PS上的苯环进行氯甲基化,然后与丙烯酸叔丁酯进行接枝[11];在SiO2表面上经(11-(2-溴-2-甲基)丙酰氧基)十一烷基三氯硅烷处理后,再用ATRP接枝制PS-b-PMA-b-PSPMA-b-PS-b-PMA三嵌段共聚物等。此外,在制备支化、星型共聚物上的应用也有很多,如用四官能引发剂C[CH2O(CH2)3-OOCCHBrCH3]4制备星型嵌段共聚物[12];制备线性和星型聚甲基丙烯酸乙二醇酯[13]。利用1,1-二苯基乙烯衍生物合成星型结构的嵌段共聚物[14];利用氧杂环丁烷制备成羟基功能化的聚氧杂环丁烷,再与2-溴异丁酰溴制备成超支化大分子引发剂(每个大分子大约有25个引发点),制备成星型支化聚合物等。
4 ATRP的展望
ATRP技术的出现开辟了活性聚合的新领域。ATRP不需要经过复杂的合成路线,因此具有十分广阔的应用前景。根据已经取得的研究成果和已经发现存在的问题,ATRP研究领域的主要研究内容集中在以下几个方面:开发新的高活性催化体系;拓宽ATRP聚合单体范围,使大部分适用于普通自由基聚合的单体能进行ATRP反应;以水为介质的ATRP技术;降低反应体系温度,使聚合反应低温化;探索简单廉价的聚合工艺,使ATRP有更广泛的适应性。由于ATRP在新材料合成方面的巨大潜力,不仅引起了学术界的关注,也引起了工业界的极大兴趣。发展高活性催化剂或发展经济实用的催化体系回收技术是ATRP工业化的关键。
[1] 唐新德,范星河,陈小芳,等.化学进展[J].2009,17(6):1089~1095.
[2] 华静,荆雄科,陈滇宝.化工科技[J].2001(4):52~56.
[3] 华静,张一峰,陈滇宝.高分子材料科学与工程[J].2002(5):21~24.
[4] 李晓林,计剑,沈家骢.高等学校化学学报[J].2005,2(2):388~392.
[5] Keary C,Zhang A,et al.Polym.Int.,[J].2002,51(7):647~652
[6] Chen XP,Padias AB,Hall HK.J.Polym.Sci.,Part A:Polym.Chem.[J].2002,40(12):1929~1936.
[7] T sarevsky NV,Sarbu T,Gobelt B,et al.Macromolecules,[J].2002, 35(16):6142~6148.
[8] Li Y,Shi P,Pan C.Macromolecules,[J].2004,37:5190~5195
[9] CiangaI,HepuzerY,YagciY.Polymer,[J].2002,43(8):2141~2149.
[10]Audouin F,Nunige S,Nuffer R,et al.Synthetic.Met.,[J].2001, 121(1~3):1149~1150.
[11]Ning FL,JiangM,MuM F,et al.J.Polym.Sci.,Part A:Polym.Chem.[J].2002,40(9):1253~1266.
[12]PanCY,TaoL,LiuY.ChinJ.Polym.Sci.,[J].2002,20(4):353~360.
[13]WangX,ZhangH,ZhongG,etal.Polymer,[J].2004,45:3637~3642
[14]DumasP,DelaiteC,HurtrezG.Macromol.Symp.[J].2002,183:29~33.
book=2010,ebook=145
10.3969/j.issn.1008-1267.2010.04.004
O621.12
A
1008-1267(2010)04-010-03
2009-10-20
猜你喜欢
功能高分子学报(2022年5期)2022-10-19
中国特种设备安全(2022年7期)2022-10-09
功能高分子学报(2022年4期)2022-08-05
劳动保护(2021年5期)2021-05-19
小资CHIC!ELEGANCE(2019年21期)2019-07-02
纺织科技进展(2016年3期)2016-11-29
橡胶工业(2016年5期)2016-02-24
中国资源综合利用(2016年6期)2016-01-22
中国塑料(2015年1期)2015-10-14
材料研究与应用(2015年4期)2015-08-23
