
Triptolide Inhibits Cell Growth and Induces G0- G1 Arrest by Regulating P21wap1/cip1 and P27 kip1 in Human Multiple Myeloma RPMI-8226 Cells
2010-07-18 11:46:07YuanLiuLinglanZengYanChenFeiZhaoRuiLiChunZhangLuWen
Yuan Liu, Ling-lan Zeng, Yan Chen, Fei Zhao, Rui Li, Chun Zhang, Lu Wen
Institute of Hematology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China.
Triptolide Inhibits Cell Growth and Induces G0- G1 Arrest by Regulating P21wap1/cip1 and P27 kip1 in Human Multiple Myeloma RPMI-8226 Cells
Yuan Liu, Ling-lan Zeng*, Yan Chen, Fei Zhao, Rui Li, Chun Zhang**, Lu Wen
Institute of Hematology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China.
Objective:To investigate the effects of triptolide(TPL) on cell growth, cell cycle and the expressions of p21wap1/cip1 and p27kip1.
Methods:MTT assay was used to determine the cell viability after triptolide treatment in human multiple myeloma RPMI-8226 cells. The effect on cell cycle distribution was determined by flow cytometry. Semi-quantitative reverse transcription-PCR was used to examine the mRNA expressions of p21wap1/cip1 and p27kip1. The protein expressions of p21 wap1/cip1 and p27kip1 were determined by Western blot.
Results:Triptolide of varying concentrations induced cell viability inhibition in dose- and time-related fashion and caused G0- G1phase arrest of cell cycle progression in RPMI-8226 cells. These effects accompanied with up-modulation of the expressions of p21 wap1/cip1 and p27kip1.
Conclusion:These results suggest that triptolide inhibit cell proliferation and cell cycle progression via up-regulating p21wap1/cip1 and p27kip1 and triptolide may exert its anti-cancer activity through this pathway.
Triptolide; RPMI-8226 cells; Cell cycle; Multiple myeloma
INTRODUCTION
Triptolide(TPL), a purified diterpenoid isolated from Chinese Herb, Tripterygium Wilfordii Hook. F, has been shown to exert both immunosuppressive and anti-inflammatory activities. Recently, triptolide has been found with remarkable anti-tumor activities in a broad range of cell culture systems andin vitrotumor models, including leukemia, colon carcinoma, gastric cancer, pancreatic cancer and breast cencer[1-5], while the molecular mechanism is not fully elucidated.
Despite the broad anti-cancer potential of triptolide, little information is so far available with regard to its effect on cell cycle in multiple myelomaRPMI-8226 cells. Cell cycle progression in eukaryotic cells is intricately regulated by the activity of cyclin and cyclin-dependent kinase (CDK) complexes. Cyclin-CDK’s activities are tightly regulated by cyclin-dependent kinase inhibitor (CDKI) which through association with cyclin-CDK complexes inhibits cell cycle progression and may function as tumor suppressor. P21wap1/cip1 and P27 kip1, members of the Cip/Kip family of CDKI, were identified as negative cell cycle regulators and mediating G0-G1arrest[6.7]. Overexpressions of p21 wap1/cip1 and p27 kip1 genes and the corresponding proteins probably play a significant role in G0-G1phase arrest and cell growth inhibition[8], which can induce cells apoptosis and exert anti-tumor activity.
In this study, we investigated the mechanism of triptolide action, and demonstrated that it induced dose- and time-dependent growth inhibition and a prolonged G0- G1phase duration. These events coincided with increased gene and protein levels ofP21wap1/cip1 and P27 kip1.
MATERIALS AND METHODS
Cell Culture
The RPMI-8226 cell line was provided by the lab of the immunology, Tongji Medical College, Huazhong University of Science and Technology (Wuhan, China). The cells were grown in RPMI-1640 with 10% fetal bovine serum, 100U/ml penicillin and 100U/ml streptomycin. Cultures were maintained in a humidified atmosphere with 5% CO2at 37°C.
Chemicals and Antibodies
Triptolide was obtained from Sigma (USA) with a purity of more than 98% and dissolved in dimethyl sulfoxide (DMSO) to prepare 1μmol/L stock solution. MTT, Propidium Iodide (PI) and DMSO were bought from Sigma. Heat-inactivated fetal bovine serum (FBS) was bought from Gibco (USA). Trizol was bought from Invitrogen(USA) and RT-PCR kit from Fermentas(USA). Monoclonal rabbit antibody against P21waf1/cip1 and polyclonal rabbit antibody against p27Kip1 were purchased from Cell Signaling(USA). Rabbit anti-human β-actin and Horseradish peroxidase (HRP)-conjugated secondary antibodies (goat anti-rabbit antibody and goat anti- mouse antibody) were bought from Santa Cruz(USA).
MTT Assay
Effect of triptolide on cell proliferation was determined by MTT assay. Briefly, 1×105RPMI-8226 cells were seeded in 96-well culture plates. After exposure to varying concentrations of triptolide (0, 30, 60, 120nmol/L) for 24, 36 and 48h , 20μl MTT solution (5mg/ml in PBS) was added to each well, and incubated for 4h at 37°C. DMSO was then added and the optical density (OD) at 570nm was measured with a 96-well multiscanner autoreader (Biotech Instruments, NY, USA). The following formula was used∶ cell proliferation inhibited (%) = [1-(OD of the experimental samples/OD of the control)] × 100%. The inhibitive effect of triptolide was assessed as IC50in each treatment group.
Cell Cycle Analysis
RPMI-8226 cells were synchronized for 24h and then incubated with varying concentrations of triptolide (0, 30, 60 or 120nmol/L) in complete medium for 24h. The cells were then harvested, washed with cold PBS, and fixed in 75% ethanol at 4°C overnight. The next day, the cells were washed and incubated with PI containing 0.05% RNase for 30min at room temperature away from light. The cell cycle distribution of the cells was then determined using FACSC alibur instrument (BD, San Diego, CA, USA).
Semiquantitative Reverse Transcription-PCR (RTPCR)
Total RNA was extracted from RPMI -8226 cells treated with or without triptolide using Trizol reagent. After quantification by spectrophotometry, the firststrand cDNA was synthesized from 2μg of total RNA with the Revert Aid First-Strand cDNA Sythesis Kit. The primers for P21 waf1/cip1 were forward 5'GGATGTCCGTCAGAACCCA3', reverse 5'CAGGT CCACATGGTCTTCC3', with a PCR product of 399bp, and the polymerase chain reaction was carried out at 94°C for 5min, followed by 35 cycles of 94°C for 50s, 55°C for 50s, 72°C for 50s, with a final extension at 72°C for 5min. P27 kip1 gene was detected with the following primers∶ forward 5'CAGCTTGCCCGAGTTCTACT3', reverse 5'TTGC AGGTCGCTTCCTTATT3', with a PCR product of 224bp fragment, and the polymerase chain reaction was carried out at 94°C for 5min, followed by 30 cycles of 94°C for 30s, 59°C for 30s, 72°C for 30s, with a final extension at 72°C for 10min. For β-actin the primers were forward 5'CCTAGAAGCATTTG CGGTGG3', reverse 5'GACTACGAGCTGCCTGAC G3', with an expected PCR product of 416bp, and the polymerase chain reaction was carried out at 94°C for 5min, followed by 30 cycles of 94°C for 30s, 60°C for 30s, 72°C for 30s, with a final extension at 72°C for 7min. The products were electrophoresed on 2% agarose gel and the ratio between the target gene and β-actin gene band density was used for quantitative evaluation.
Western Blot Analysis
For preparation of total cell lysates, cells were collected and lysed in lysis buffer (50mmol/L HEPES, 150mmol/L NaCl, 1% Triton X-100, 5mmol/L EGTA, 50mmol/L-glycerophosphate, 20mmol/L NaF, 1mmol/L Na3VO4, 2mmol/L phenylmethyl sulfonyl fluoride, 10μg/ml leupeptin and 10μg/ml aprotinin) by incubating on ice for 1h. The lysates were then centrifuged at 12000g for 15min at 4°C. The supernatant was collected and the total protein concentrations were determined using the BCA assay by spectrophotometer (Biotech Instruments, NY, USA). The samples were separated on 8%-12%SDS-PAGE and transferred onto NC membranes. After blocking with 5% non-fat dry milk in blocking buffer (25mmol/L Tris, pH 7.5, 150mmol/L NaCl, and 0.1% Tween 20), the membranes were incubated with primary antibodies at 4°C overnight. The membranes were then incubated with appropriate peroxidaseconjugated secondary antibodies, and the protein expression was detected by ECL substrate solution (Themo, USA). Densitometric analysis was performed using Quantity One software.
Statistical Analysis
The statistical significance of difference between control and treatment groups was determined by the Student'st-test using SPSS 17.0. Values were shown as ¯x±s,P<0.05 was considered statistically significant.
RESULTS
Effect of Triptolide on Proliferation of RPMI-8226 Cells
The effect of triptolide on the proliferation of RPMI-8226 cells was determined using MTT assay. As shown in Figure 1, triptolide (0, 30, 60, 120 nmol/L) treatment resulted in a significant dose- and time-dependent inhibition of cell growth, the IC50value for 36h was 121.76±2.01 nmol/L. Triptolide treatment also resulted in time-dependent reduction in cell proliferation, this effect was more pronounced at 48h post-treatment (Figure 1). A marked effect on cell growth inhibition was observed with gradually increased exposure concentration and time.

Figure 1. Effect of triptolide on the proliferation of RPMI-8226 cells by MTT assay. The cells were treated with different concentrations of triptolide for 24, 36 and 48h. The values were represented as the cell viability inhibition rate and each data represent as¯x±sof four independent experiments. (n=4,P<0.05)
Effect of Triptolide on Cell Cycle in RPMI-8226 Cells
The effect of triptolide on cell cycle distribution was evaluated by flow cytometry. Compared with control group (37.80% cells in G0-G1phase), triptolide treatment resulted in an appreciable arrest of cells in G0-G1phase of cell cycle after 24h. The treatment caused an arrest of 48.60% cells in G0-G1phase of cell cycle at of 30nmol/L, increased to 50.33% at 60nmol/L, and further to 61.33% at the concentration highest dose of 120nmol/L in RPMI-8226 cells (Figure 2). This increase in G0-G1cell population accompanied with a concomitant decrease of cells in S phase, while the proportion of G2-M phase cells had no significant change (Figure 2).
Effect of Triptolide on the mRNA Expressions of p21 Waf1/Cip1 and p27 Kip1 in RPMI-8226 Cells
We examined the gene expression of p21 Waf1/Cip1 and p27 Kip1 in RPMI-8226 cells by RTPCR. As shown in Figure 3, the mRNA relative quantification of P21wap1/cip1 and P27kip1 revealed a significant dose-dependent increase in RPMI- 8226 cells treated with varying concentrations of triptolide (0, 30, 60 and 120nmol/L) for 48h. RT-PCR analysis revealed that triptolide treatment results in appreciable increase of gene expression of P21wap1/ cip1and P27 kip1 even at the lowest concentration of 30nmol/L, with an obvious increase at the highest concentration of 120nmol/L (Figure 3)
Effect of Triptolide on the Protein Expressions of P21wap1/cip1 and P27 kip1 in RPMI-8226 Cells
The effect of triptolide on the expression of P21 wap1/cip1 and P27 kip1 was also observed in the protein levels by Western blot. Similar to the outcome of RT-PCR, as shown by Western blot analysis, triptolide treatment led to significant dose-dependent up-regulation of the protein expressions of P21wap1/cip1 and P27 kip1 with maximum at the highest concentration at 120nmol/L (Figure 4). The protein density of P21wap1/cip1 and P27 kip1 increased gradually along with the exposure concentrations. The change in protein expression was not due to differences in the amount of protein loaded on the gels as the equivalent protein loading was confirmed by β-actin as shown (Figure 4).
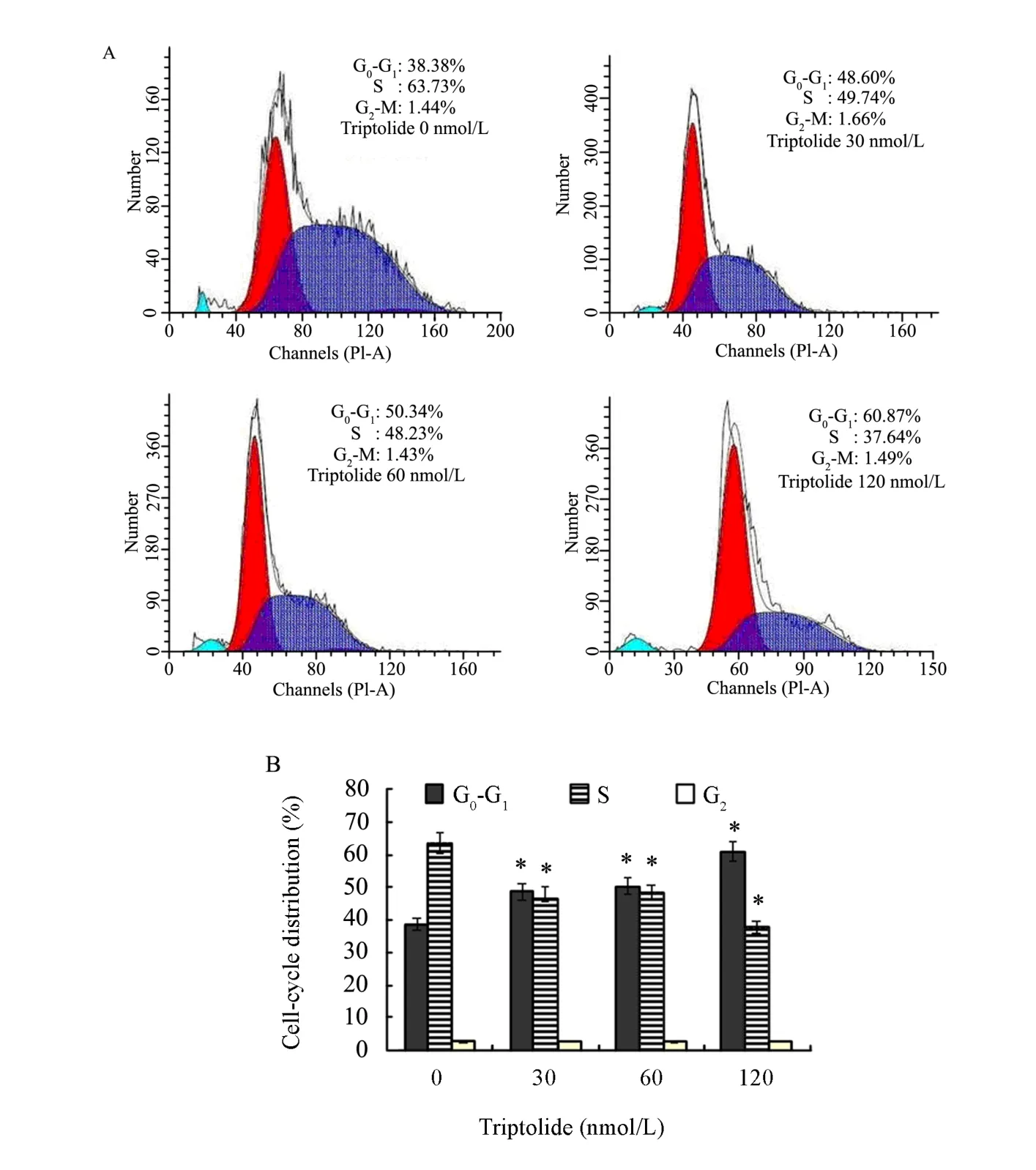
Figure 2. Induction of cell cycle arrest by triptolide in RPMI-8226 cells. Cells were exposed to specific concentrations of triptolide for 24h and subjected to flow cytometry to analyze the cell cycle distribution, as detailed in the Materials and Methods. (A) Cell cycle distribution was detected by PI assay; (B) Data was summarized and presented as¯x±sof three independent experiments. (n=3,*P<0.05)

Figure 3. Effects of triptolide on the mRNA expressions of P21wap1/cip1 and P27 kip1. (A) Line 1∶ negative control untreated with triptolide; Line 2, Line 3 and Line 4∶ cells treated with triptolide at 30nmol/L, 60nmol/L and 120nmol/L for 48 h, respectively. (B) Gene expression was semiquantified by relative densities of P21wap1/cip1 and P27 kip1 to the density of β-actin. Data represented as¯x±sof at least three independent experiments (n=3,*P<0.05).

Figure 4. Effects of triptolide induced the protein expression of P21wap1/cip1 and P27 kip1 in RPMI- 8226 cells. (A) Line 1∶ negative control untreated with triptolide; Line 2, Line 3 and Line 4∶ cells treated with triptolide of 30nmol/L, 60nmol/L and 120nmol/L for 48 h respectively; (B) The values represent as change in protein expression of the bands normalized to β-actin (n=3,*P<0.05).
DISCUSSION
Previous studies have reported that triptolide could inhibit proliferation and induce apoptosis in tumor cells[10,11], and it has been suggested that triptolide induced apoptosis by alternating pathways involving p53 and PI3K/Akt[12,13], and by inducing caspase-dependent cell death via mitochondrial pathway10]. So far there is no available information about the anti-tumor effect of triptolide related with cell cycle in human multiple myeloma RPMI-8226 cells. We investigated the mechanism of the reduction in cell viability and cell cycle arrest in RPMI-8226 cells by triptolide in our study.
Ourin vitrodata demonstrated that treatment of RPMI-8226 cells with triptolide induced inhibition of proliferation in a dose- and time-dependent manner and G0-G1phase arrest of RPMI-8226 cells in a dose-dependent manner. We, therefore, hypothesized that triptolide-mediated cell growth inhibitory and cell cycle arrest might perturb cell growth, thus exerted anti-tumor activity. Based on preliminary assays, we next evaluated the mechanism by which triptolide induces G0-G1phase arrest in RPMI-8226 cells. Our data suggested that triptolide-induced increases of the expressions of P21wap1/cip1 and P27kip1 have a role in the blockade of RPMI-8226 cells in G0-G1phase and growth inhibition.
Agents that can modulate apoptosis may be able to affect steady-state cell proliferation, which may be useful in the management and therapy of cancer[14,15]. In recent years the interest of several investigators has been focused on cell cycle regulatory machinery considering that the distribution of normal cell cycle is a critical step in cancer development. Cellular proliferation is regulated by a balance between proliferation, cell cycle arrest and programmed cell death. Disorder of cell cycle is closely related to the progression and aggressiveness of cancers11. Cell cycle arrest provides an opportunity for cells to undergo either repair mechanisms or apoptosis. The major regulatory events leading to cell proliferation and differentiation occur within G0-G1phase of cell cycle, when the cell commits itself to DNA replication and both positive and negative external signals are integrated into the cell cycle. G0-G1checkpoint abrogation is a common phenomenon in carcinogenesis which endows the tumor cells with limitless replicative potential13. G0-G1to S phase transition plays a crucial role in maintaining the genomic integrity because this phase is critically linked to external stimuli and also commits the cells to DNA replication and subsequent mitosis[16,17]. In most of the malignancies, cancer cells become resistant to apoptosis and/or do not respond to the cytotoxic effects of chemotherapeutic agents. It has been recognized that the control of cell cycle progression in cancer cells is an effective strategy to halt tumor growth[12,13], as the molecular analyses of cancers have revealed that cell cycle regulators are frequently deregulated in most common malignancies[18,19]. Our data demonstrated that treatment of RPMI-8226 cells with triptolide induced G0-G1phase arrest of cell cycle progression, indicating that one of the mechanisms by which triptolide inhibited the proliferation of RPMI-8226 cells was the inhibition of cell cycle progression.
Cell cycle progression is governed by the well-orchestrated activation and inactivation of cyclins, CDKs and CDKI. Gene abnormalities andaberrant expressions of cell cycle regulators play a pivotal role in the tumors, of which CDKI are major regulators. P21wap1/cip1 and P27kip1 are universal members of CDKI, whose activity is a keen cause of cancer progression, and commonly up-regulated in response to antiproliferative signals[20]. It has been found that P21wap1/cip1 and P27 kip1 associated with cyclin-CDK that inhibit the phosphorylation of CDK, a crucial event for G0- G1phase to S transition. P21wap1/cip1 is a downstream effecter of p53 and belongs to Cip1/Kip1 of CDKI. P21wap1/cip1 expression can bind with cyclinD1 and cyclinE through a p53-independent pathway and influence the functional status at p53 which is a transcription factor able to stop cell cycle of G0- G1phase and to induce apoptosis[21]. Thus it is a potential tumor suppressor, and likely plays an important role in tumor therapy development. P27kip1, also a potential tumor suppressor, has been found to be deregulated in a series of human cancer[22]. P27kip1 is believed to primarily regulate the progression of cells from late G0- G1to S phase by interacting with cyclinE-CDK2 complexes. Elevated expression of P27kip1 leads to G0- G1arrest in many cell lines[20]. There are evidences demonstrating that high levels of P27kip1 lead to growth arrest as well as enhancement of apoptosis in tumor cells[23]. Numerous studies showed that both P21wap1/cip1 and P27 kip1 regulate the progression of cells in G0- G1phase and induction of these molecules causes a blockade of G1→S transition thereby resulting in G0-G1phase arrest, a further inhibition of proliferation[24]. Thus, activating the G0-G1checkpoint by up-regulating the expression of P21wap1/cip1 and P27 kip1 is a logical approach for controlling cancer cells proliferation[25]. Consistent with this notion, our study showed that increase of P21wap1/cip1 and P27 kip1 was a significant event in triptolide-induced G0- G1arrest and inhibition of proliferation in RPMI-8226 cells. A significant increase in mRNA and protein expressions of P21wap1/cip1 and P27 kip1 by triptolide suggest their potential in inhibition of human multiple myeloma cells.
In conclusion, our study indicates that triptolide inhibits cell growth and induces G0- G1phase arrest of human multiple myeloma RPMI-8226 cells. The data also provide evidences that triptolide-induced cell cycle arrest and the inhibition of proliferation in RPMI-8226 cells are mediated through the upregulation of P21wap1/cip1 and P27 kip1. This observation is of particular significance in suggesting triptolide as a promising agent for prevention and therapy of multiple myeloma and CDKI may be therapeutic targets that should be considered in the future treatment of multiple myeloma.
Acknowledgment
This work was supported by a grant from the Department of Immunology, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
REFERENCES
[1] Lou YJ, Jin J. Triptolide down-regulates bcr-abl expression and induces apoptosis in chronic myelogennous leukemia cells[J]. Leuk Lymphoma 2004; 45∶ 373-6.
[2] Tang XY, Zhu YQ, Tao WH, et al. Synergistic effect of triptolide combined with 5-fluorouracil on colon carcinoma[J]. Postgrad Med J 2007; 83∶ 338-43.
[3] Jan HJ, Lee CC, Lin YM, et al. Rosiglitazone reduces cell invasiveness by inducing MKP-1 in human U87MG glioma cells[J]. Cancer Lett 2009; 277∶141-8.
[4] Liang M, Fu J. Triptolide inhibits interferon-gammainduced programmed death-1-ligand 1 surface expression in breast cancer cell[J]. Cancer Lett 2008; 270∶ 337-41.
[5] Philips PA, Dudeja V, McCarroll JA, et al. Triptolide induces pancreatic cancer cell death via inhibition of heat shock protein 70[J]. Cancer Res 2007; 67∶9407-16.
[6] Roy S, Gu M, Ramasamy K, et al. p21/Cip1 and p27/Kip1 are essential molecular targets of inositol hexahosphate for its antitumor efficacy against prostate cancer[J]. Cancer Res 2009; 69∶ 1166-73.
[7] Baiz D, Pozzato G, Dapas B, et al. Bortezomib arrests the proliferation of hepatocellular carcinoma cells HepG2 and JHH6 by differentially affecting E2F1, p21 and p27 levels[J]. Biochimie 2009; 91∶ 373-82.
[8] Besson A, Dowdy SF, Roberts JM. CDK inhibitors∶cell cycle regulators and beyond[J]. Dev Cell 2008; 14∶ 159-69.
[9] Gao F, Yi J, Yuan JQ, et al. The cell cycle related apoptotic susceptibility to arsenic trioxide is associated with the level of reactive oxygen species[J]. Cell Res 2004; 14∶ 81-5.
[10] Carter BZ, Mak DH, Schober WD, et al. Triptolide induces caspase-dependent cell death mediated via the mitochondrial pathway in leukemia cells[J]. Blood 2006; 108∶ 630-7.
[11] Krug U, Ganser A, Koeffler HP. Tumor suppressor genes in normal and malignant hematopoiesis[J]. Oncogene 2002; 21∶ 3475-95
[12] Zanetti M, Stocca A, Dapas B, et al. Inhibitory effects of fenofibrate on apoptosis and cell proliferation in human endothelial cells in high glucose[J]. J MolMed 2008; 86∶ 185-95.
[13] Singh RP, Agarwal R. Natural flavonoids targeting deregulated cell cycle progression in cancer cells[J]. Curr Drug Targets 2006; 7∶ 345-54.
[14] Athar M, Back JH, Kopelovich L, et al. Multiple, molecular targets of resveratrol∶ anti-carcinogenic mechanisms[J]. Arch Biochem Biophys 2009; 486∶95-102.
[15] Coqueret O. New roles for p21 and p27 cell-cycle inhibitors∶ a function for each cell compartment[J]? Trends Cell Biol 2003; 13∶ 65–70.
[16] Tian Y, Song Y. Effects of inositol hexaphosphate on proliferation of HT-29 human colon carcinoma cell line[J]. World J Gastroenterol 2006; 12∶ 4137-42.
[17] Roy S, Singh RP, Aqarwal C, et al. Downregulation of both p21/Cip1 and p27/kip1 produces a more aggressive prostate cancer phenotype[J]. Cell Cycle 2008; 7∶ 1828-35.
[18] Enders GH, Maude SL. Traffic safety for the cell∶influence of cyclin-dependent kinase activity on genomic stability[J]. Gene 2006; 371∶ 1-6.
[19] Schwartz GK, Shah MA. Targeting the cell cycle∶ a new approach to cancer therapy[J]. J Clin Oncol 2005; 23∶ 9408–21.
[20] Zheng JY, Wang WZ, Li KZ, et al. Effect of p27(kip1) on cell cycle and apoptosis in gastric cancer cells[J]. World J Gastroenterol 2005; 11∶ 7072-7.
[21] Laochim E, Michael M, Stavropoulos NE, et al. Expression patterns of cyclinD1, E and cyclindependent kinase inhibitors p21wap1/Cip1 and p27/Kip1 in Urothelial carcinoma∶ Correlation with other cell-cycle-related proteins (Rb, p53, Ki-67 and PCNA) and clinicopathological features[J]. Urol Int 2004; 73∶ 65-73.
[22] Korkolopoulou P, Christodoulou P, Konstantinidou AE, et al. Cell cycle regulators in bladder cancer∶ A multivariate survival study with emphasis on p27kip1[J]. Hum Pathol 2000; 31∶ 751-60.
[23] Katner AL, Hoang QB, Gootam P, et al. Induction of cell cycle arrest and apoptosis in human prostate carcinoma cells by a recombinant adenovirus expressing P27kip1[J]. Prostate 2002; 53∶ 77-87.
[24] Onumah OE, Jules GE, Zhao Y, et al. Overexpression of catalase delays G0- G1 to S phase transition during cell cycle progression in mouse aortic endothelial cells[J]. Free Radic Biol Med 2009; 46∶ 1658-67.
[25] Roy S, Kaur M, Aqarwal C, et al. p21 and p27 induction by silibinin is essential for its cellcycle arrest effect in prostate carcinoma cells[J]. Mol Cancer Ther 2007; 6∶ 2696-707.
R733.3 Document code: A Article ID: 1000-9604(2010)02-0141-07
10.1007/s11670-010-0141-5
2009−10−22; Accepted 2010−01−16
This work was supported by the National Natural Science Foundation of China(No. 30700882)
*Contributed equally to this study.
**Corresponding author.
E-mail∶ zhangchun23@yahoo.com.cn
© Chinese Anti-Cancer Association and Springer-Verlag Berlin Heidelberg 2010
 Chinese Journal of Cancer Research2010年2期
Chinese Journal of Cancer Research2010年2期
- Chinese Journal of Cancer Research的其它文章
- Different Outcome of Myeloid Sarcoma with Spinal Cord Compression Preceding Acute Myeloid Leukemia: Report of Two Cases and Review of Literatures
- Effects of Triptolide on Histone Acetylation and HDAC8 Expression in Multiple Myeloma in vitro
- CGI-100 Specific shRNA Inhibits Proliferation and Induces Differentiation in Leukemia K562 Cells
- Expression of Embryonic Stem Cell Marker Oct-4 and Its Prognostic Significance in Rectal Adenocarcinoma
- Differential Diagnosis of Warthin's Tumor Complicated with Lung Adenocarcinoma by 18F- FDG PET/CT Imaging and Radioisotope Scanning with Tc-99m Pertechnetate: A Case Report and Literature Review
- Synergistic Action of fMLP-boanmycin Combination on the Growth of Mouse Colon Carcinoma and Its Action Mechanisms