
1-S-二苯基膦-2-R-二(3,5-二甲基苯基)膦二茂铁的合成
2010-05-29 05:45申永存徐维赟
武汉工程大学学报 2010年5期
申永存,徐维赟
(武汉理工大学化学工程学院制药工程系,湖北 武汉 430070)
0 引 言
自上世纪70年代以来,不对称催化氢化一直是国内外科学工作者关注的焦点和研究的热点[1-4].在不对称催化中,双膦配体是一种常用的催化剂配体,大多数双膦配体易被氧化,而二茂铁类双膦配体则相对稳定,已被广泛应用于大规模不对称催化氢化反应中[5-6].1-S-二苯基膦-2-R-二(3,5-二甲基苯基)膦二茂铁((R)-(S)-PPF-Pxyl2)(1)是其代表之一,瑞士先正达公司已成功用于除草剂的规模化生产中[7].为了推动我国除草剂的发展,笔者开展该产品的工艺研究.
关于双膦配体(1)的合成,文献报道[6]以二茂铁为原料经多步合成,但是路线中采用了多次柱层析分离纯化的方法,不适用于工业化生产.为此笔者在现有文献的基础上[6-9]对手性二茂铁类双膦配体的合成方法进行改进,采用“一锅烩”的方法完成还原、酯化、胺解等反应,得到的混合物经减压蒸馏得到高纯度的N,N-二甲基胺乙基二茂铁(6),化合物(6)经拆分、二苯基膦化、膦代等步骤合成1-S-二苯基膦-2-R-二(3,5-二甲基苯基)膦二茂铁(1),实现了一种方法简单、产率高、成本低、可产业化的手性二茂铁类双膦配体1的合成方法.
本实验的技术路线是:以二茂铁(2)为原料经乙酰化、还原、酯化生成化合物乙酰氧乙基二茂铁(5),化合物(5)与二甲胺置换反应生成N,N-二甲胺乙基二茂铁(6),(6)通过手性酸拆分得2-R-N,N-二甲胺乙基二茂铁(6)和2-S-二甲胺乙基二茂铁(6), 2-R-N,N-二甲胺乙基二茂铁(6)与丁基锂反应得二茂铁的锂盐,再与二苯基氯化膦(8)反应生成占绝对优势构型的1-S-二苯基膦-2-R-N,N-二甲胺基乙基二茂铁(7),(7)与二(3,5-二甲基苯基)膦(9)反应,生成双膦配体1-S-二苯基膦-2-R-二(3,5-二甲基苯基)膦二茂铁(1).合成路线如图1所示.
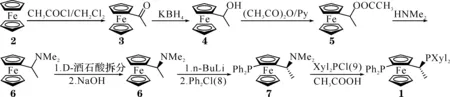
图1 1-S-二苯基膦-2-R-二(3,5-二甲基苯基)膦二茂铁的合成路线
1 实验部分
1.1 仪器与药品
Bruker Avance 500 DMX 核磁共振仪(TMS为内标,CDCl3为溶剂); Agilent 1100 Series 液相色谱仪;Agilent 6890 Series 气相色谱仪;日本分光株式会社Jasco P1010旋光仪;北京泰克有限公司 X-4型数字显示熔点仪.
二苯基氯化膦(8)与二(3,5-二甲基苯基)膦(9)均从北京百灵威化学试剂有限公司购得,其他药品及试剂均为市售化学纯或工业级药品.
1.2 实验方法
1.2.1 2-N,N-二甲胺乙基二茂铁(6)的合成 向装有机械搅拌、回流冷凝管、滴液漏斗的250 mL的三口瓶中加入18.6 g(0.1 mol)二茂铁(2)、10 g(0.12 mol)氧化锌、60 mL二氯甲烷,开始缓慢滴加7.8 mL(0.11 mol)乙酰氯,加毕,室温下反应2 h后将反应液水洗,水层用50 mL二氯甲烷萃取两次,有机层,干燥、浓缩,向残留物中加入5.4 g(0.1 mol硼氢化钾, 25 mL甲醇,升温至60 ℃反应,TLC 跟踪反应原料消失即毕,减压回收甲醇,向残留物中加入14 mL(0.1 mol)醋酐、16 mL吡啶,室温搅拌反应,TLC跟踪反应原料消失即毕,减压回收醋酐及吡啶, 向剩余物中再加入40 mL甲醇和41 mL33%(0.3 mol)的二甲胺水溶液,室温搅拌反应4 h,反应体系用乙酸乙酯和水萃取,浓缩回收溶剂,残留物减压蒸馏收集110 ℃(0.07 kPa)馏分约20.5 g (6),收率80%,GC分析含量约不低于99%.1H NMR(CDCl3):δ4.08~4.03(m,9H), 3.54(q, J=6.9Hz,1H), 2.02(s,6H), 1.38(d,J=7.2 Hz,3H);13C NMR(CDCl3):δ87.5,69.8,69.0,67.8,67.6,67.3,59.1,41.0,16.5.
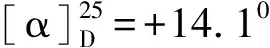

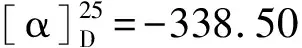
2 结果与讨论
本实验以二茂铁为原料经付克酰化反应得到乙酰二茂铁,研究了氯化铝、氯化锌、三氟化硼的乙醚溶液等路易斯酸催化下的乙酰化反应,结果表明氧化锌最理想,付克酰化反应收率可达95%以上,而在其他路易斯酸催化下,容易产生二取代的杂质,其收率也仅为35%~80%.
乙酰二茂铁经硼氢化钾还原、醋酐酯化、二甲胺氨解等反应合成化合物(6),可在同一反应器中完成,无需经柱层析纯化,减少了操作步骤、提高了反应效率,收率达到80%左右.
3 结 语
本实验对手性双膦配体1-S-二苯基膦-2-R-二(3,5-二甲基苯基)膦二茂铁的合成工艺进行改进,简化了操作,保证了产品质量,为工业化生产提供了切实可行的技术支持,为除草剂在我国的规模化生产提供了保证.
参考文献:
[1]宋庆宝,东宇.二茂铁手性膦配体研究的一些进展[J].有机化学,2007,27(1):66-71.
[2]Arrayas R G, Adrio J, Carretero J C. Recent App-lications of Chiral Ferrocene Ligands in Asymmetric Catalysis[J]. Angew Chem Int Ed,2006,45(46):7674-7715.
[3]Manuel A F, Shen Q L, Hartwig J F. Highly Efficient and Functional Group Tolerant Catalysts for the Palladium-Catalyzed Coupling of Aryl Chlorides with Thiols[J].Chem Eur J,2006,12(30):7782-7796.
[4]Colacot T J. A Concise Update on the Applications of Chiral Ferrocenyl Phosphines in Homogeneous Catalysis Leading to Organic Synthesis[J].Chem Rev,2003,103(8):3101-3118.
[5]Feng X D, Pugin B, Blaser H U,et al. Josiphos Ligands with an Imidazolium Tag and their Application for the Enantioselective Hydrogenation in Ionic Liquids[J]. Adv Synth Catal,2007,349(10):1803-1807.
[6]Hayashi T, Mise T, Yamamoto K. Asymmetric Synthesis Catalyzed by Chiral Ferrocenylphosphine-Transition Metal Complexes. I. Preparation of Chiral Ferrocenylphosphines[J]. Bull Chem Soc Jpn,1980,53(4):1152-1156.
[7]Dorta R, Broggini D, Stoop R. Chiral Xyliphos Complexes for the Catalytic Imine Hydrogenation Leading to the Metolachlor Herbicid:Isolation of Catalyst Substrate Adducts[J]. Chem Eur J,2004,10(1):267-278.
[8]Togni A, Breutel C Schnyder A, A Novel Easily Accessible Chiral Ferrocenyldiphosphine for Highly Enantioselective Hydrogenation, Allylic Alkylation, and Hydroboration Reactions.[J].J Am Chem Soc,1994,116(9):4062-4066.
[9]Tappe K, Knochel P. New Efficient Synthesis of Taniaphos Ligands: Application in Ruthenium-and Rhodium-Catalyzed Enantioselective Hydrogenations.[J].Tetrahedron:Asymmetry,2004,15(1):91-102.
[10]Marquarding D, Klusacek H, George G, et al. Correlation of Central and Planar Chirality in Ferrocene Derivatives [J].J Am Chem Soc,1970,92(8):5389-5393.
[11]Blaser H U, Brieden W, Pugin B, et al. Solvias Josiphos Ligands: from Discovery to Technical Applications[J]. Topics in Catalysis,2002,19(1):3-16.
猜你喜欢
国际放射医学核医学杂志(2020年2期)2020-05-30
山东化工(2019年7期)2019-04-27
中国资源综合利用(2017年1期)2018-01-22
中国塑料(2017年2期)2017-05-17
固体火箭技术(2016年5期)2016-11-03
中国塑料(2015年5期)2015-10-14
遵义医科大学学报(2014年1期)2014-09-10
中国药业(2014年17期)2014-05-26
无机化学学报(2014年9期)2014-02-28
郑州大学学报(理学版)(2013年3期)2013-03-11
