
流化床光催化臭氧氧化苯酚动力学模型
2010-03-14 06:39董双石周丹丹任南琪张静仁于丽丽
哈尔滨工业大学学报 2010年10期
董双石,周丹丹,任南琪,张静仁,于丽丽
(1.哈尔滨工业大学市政环境工程学院,哈尔滨150090,dongshuangshi@gmail.com;
2.吉林大学环境与资源学院,长春130023;3.中国市政工程东北设计研究院,长春130021)
工业废水中的有毒有害污染物导致了严重的水体污染问题.某些废水的可生物降解性较差,而采用传统的化学或物化方法通常会产生二次污染,因此,探寻一种高效率、低成本的处理方法十分必要.近年来,光催化氧化技术在去除水中污染物领域有了显著进展[1-5].其工作原理为:当半导体材料受到能量大于或等于能隙的光时,产生空穴-电子对.水中的溶解氧作为俘获剂,抑制了电子和空穴的复合,使得空穴能够氧化表面吸附的H2O或OH-离子生成具有强氧化性的羟基自由基,进而氧化水中的污染物.很多研究均已证实,如果采用臭氧作为俘获剂能够进一步抑制电子和空穴的复合,提高羟基自由基的产生量,而且臭氧本身也有较强的氧化作用[6-8].因此,光催化臭氧氧化技术可以认为是臭氧氧化、光臭氧氧化以及光催化氧化的综合作用[9-10].
二氧化钛因其较高的禁带能量、化学惰性且无毒价廉,是较为常用的光催化剂[11-13].负载型二氧化钛虽然比粉末二氧化钛效率低[14],但由于不需要后续的分离而更易于实际应用.与固定床相比,流化床内催化剂与反应物接触更充分,光利用率更高[15-17].
目前国内外对流化床中光催化处理废水的研究主要集中在处理效率以及运行条件优化上,对流化床中光催化臭氧氧化研究还鲜见报道.本研究采用溶胶-凝胶法将纳米二氧化钛负载到球形活性炭上作为光催化剂,在流化床中对比了O3、O3/AC、UV/O3、UV/O3/AC以及UV/O3/TiO2-AC等5种氧化过程对苯酚废水的处理效率.建立UV/O3/AC和UV/O3/ TiO2-AC氧化过程的动力学模型,为流化床光催化反应器的设计与放大提供依据.
1 实验
1.1 负载型二氧化钛的制备与表征
将体积份数为20%的乙酰丙酮加入到1∶9的钛酸四丁酯(Aldrich,97%)与无水乙醇的混合液中,然后加入浓硝酸调节pH值至2,在剧烈搅拌条件下滴入去离子水得到黄色透明溶胶.待释放反应热后以100 g/L混合液的比例加入球形活性炭(Kureha,平均直径0.6 mm),继续搅拌2 h后过滤转至烘干箱,115℃下保持1 h.然后样品在马弗炉中500℃下煅烧2 h,升温速度控制在5℃/min.冷却至室温后超声清洗2 min.
采用扫描俄歇电子显微镜(Thermo VG Microlab 350)对样品进行SEM表征.负载到活性炭表面物质的俄歇电子能谱表明其成分为二氧化钛.采用差重法测得负载颗粒的二氧化钛质量分数为6.51%.
1.2 活性炭与负载二氧化钛的等温吸附/脱附
将精确量取一定堆积体积(5 mL)的活性炭颗粒与负载二氧化钛颗粒,分别放入250 mL三角瓶中,再分别加入100,250,500,1 000,2 000,2 500,3 500,4 000和5 000 mg/L的苯酚溶液100 mL.用玻璃瓶塞密封后在25℃恒温箱内振荡24 h后,检测水样中苯酚平衡质量浓度ρ,通过平衡法计算出吸附量q.
将上述实验中的颗粒滤出放入一定量的去离子水中,振荡24 h后检测水样中的苯酚平衡质量浓度,通过质量质量浓度守恒法计算出残留吸附量q,并由此计算出脱附量.
1.3 流化床光催化反应器
流化床光催法反应器及其降解反应工艺流程如图1所示.采用GE紫外灯(15 W)作为紫外光源,位于流化床中心轴,紫外灯外套直径为25 mm石英套管,反应器内壁(直径为127 mm)与套管外壁之间形成的环形区域为流化区,即光催化氧化反应区.反应器外壁用铝箔覆盖防止外界光源的干扰.流化床初始填充高度为90 mm,液相通过分布板后,进入反应区与催化剂接触,在紫外线作用下发生光催化降解反应.反应段中部进空气,去除气泡进入床层对光的散射作用.臭氧发生器产生的臭氧在文丘里管(Mazzei,484)的作用下被吸入到反应器中.反应段顶部安装滤网以防止颗粒流失.上部安装直径为305 mm的扩大段,对气液两相进行有效分离.水箱内安装有热交换管以保持液相温度25℃.液相总体积为15 L,其中在反应器中11.5 L.
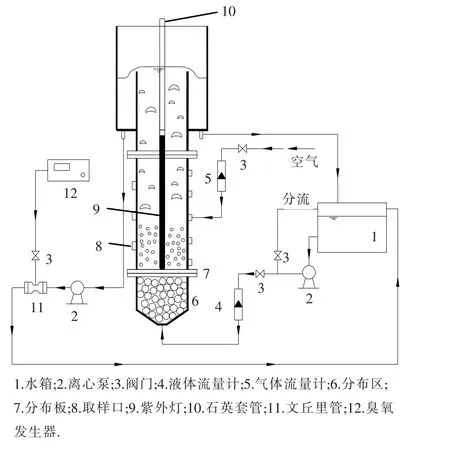
图1 流化床光催化反应器示意图
1.4 分析项目与方法
采用4-氨基安替比林法测定水中苯酚质量浓度,吸光度值由Hack Dr/4000紫外分光光度计测定.苯酚的降解率通过下式计算:
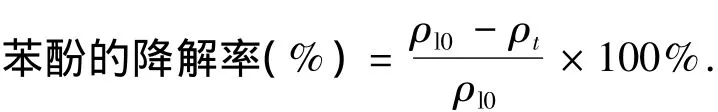
其中ρl0为液相苯酚初始质量浓度,ρt为反应t时刻的液相苯酚质量浓度.
DO与pH值由VWR Symphony PD80分析仪测定.测得液相中DO为饱和值,而实验中pH值为7.2,反应过程中基本不变.水中臭氧则以Hack AccuVac瓶取样,采用Hack Dr/4000紫外分光光度计内置程序测定,在实验期间其质量浓度在0.02 mg/L左右.
2 结果与讨论
2.1 活性炭与催化剂的吸附特性
活性炭与催化剂对苯酚的吸附与脱附结果见图2.可以看出,两种颗粒对苯酚的吸附与脱附是可逆的.与其他吸附模型相比,在稀溶液下弗兰德里希吸附等温模型对实验结果的预测更准确.其等温吸附模型方程如下:
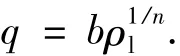
式中:q为单位堆积体积活性炭或催化剂吸附苯酚的质量浓度,mg/mL;ρl为吸附平衡时液相苯酚质量浓度,mg/L;b和n为给定吸附系统的常量.b值代表了在苯酚吸附平衡质量浓度为1 mg/ L时颗粒对苯酚的吸附总量,而1/n则与吸附反应所需能量有关,其值越高,反应所需能量越底.活性炭(b1,n1)及催化剂(b2,n2)的吸附常量通过logq-ρl的线性回归得出,分别为 b1= 21.28 mg/mL、n1=3.06,b2=20.88 mg/mL,n2=3.12.可以看出,由于负载了TiO2,催化剂颗粒的吸附能力相对于原活性炭有所下降,但并不明显,原因是负载的TiO2仅有6.51%.此部分结果将用于降解苯酚动力学模型的计算.
2.2 苯酚的降解动力学
光催化氧化有机物的反应速率一般用Langmiur-Hinshelwood(L-H)模型[18-19]进行描述.本研究选择初始苯酚质量浓度分别为6,16,34,80 mg/L以及186 mg/L时,采用了此模型预测流化床反应器中苯酚的光催化降解动力学行为.液相流速和气相流速分别为13.8,3 L/min.反应速率可以表达为
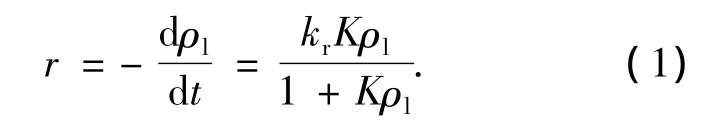
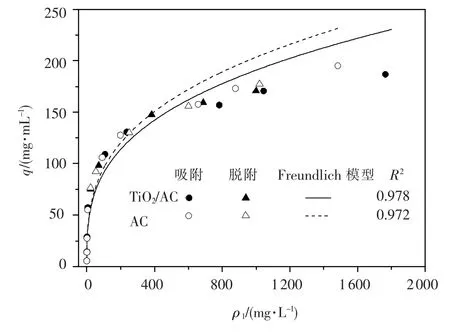
图2 活性炭(AC)与负载型二氧化钛(TiO2-AC)颗粒的等温吸附/脱附
其中kr为流化床反应器内总表观反应速率常数,K为反应物吸附常数.当初始反应速率r0与初始苯酚质量浓度ρl0已知,并代入方程(1),得
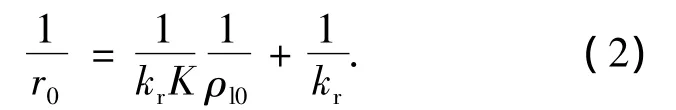
将实验数据代入方程(2)后对1/r0-1/ρl0关系进行线性描述,结果见图 3.求出 kr= 0.336 mg/(L·min),K=5.14×10-4L/g,决定系数R2=0.995,预测结果表明,L-H模型能够较好地描述制备的光催化剂在流化床中对苯酚的光催化反应动力学行为.
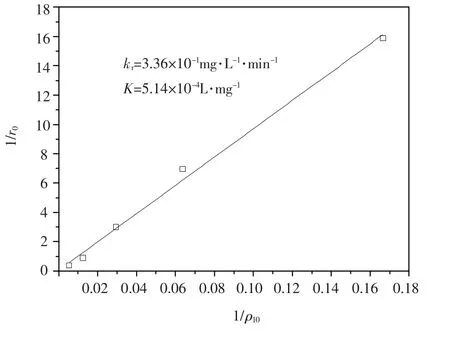
图3 流化床光催化氧化苯酚过程中初始反应速度倒数(1/r0)与初始苯酚质量浓度倒数(1/ρ10)的关系
2.3 不同氧化过程的对比
对比了流化床中以下5种氧化过程降解苯酚废水的效率:均相臭氧氧化(O3)、均相光臭氧氧化(UV/O3)、多相光臭氧氧化(UV/O3/AC)、多相光催化氧化(TiO2-AC/UV/O2)以及多相光催化臭氧氧化(TiO2-AC/UV/O3).实验采用液相苯酚初始质量浓度为(11.7±0.2)mg/L.
从图4可以看出,UV/O3氧化过程降解苯酚效率比O3过程高15%左右,这是因为UV与O3的联合作用产生了更多的·OH.多相光臭氧氧化、多相光催化氧化以及多相光催化臭氧氧化苯酚的结果表明,反应结束后液相中剩余苯酚质量浓度高于均相臭氧氧化和均相光臭氧反应结束后液相中剩余苯酚质量浓度.一方面由于在多相光臭氧氧化、多相光催化氧化以及多相光催化臭氧氧化过程中存在吸附性颗粒,一定量的·OH被吸附到颗粒表面,使液相的·OH质量浓度降低;另一方面是由于液相苯酚质量浓度的降低带来了苯酚的脱附.
表1的结果表明,UV/O3/AC与TiO2-AC/ UV/O3对苯酚的氧化过程,光催化的作用使降解苯酚效率提高了34.5%.而TiO2-AC/UV/O2过程对苯酚的降解作用并不明显.这说明在流化床光催化反应器中,O3比O2更适合作为光催化产生的空穴-电子对中电子的捕获剂.这两个过程将在下面的模型部分作详细的对比.
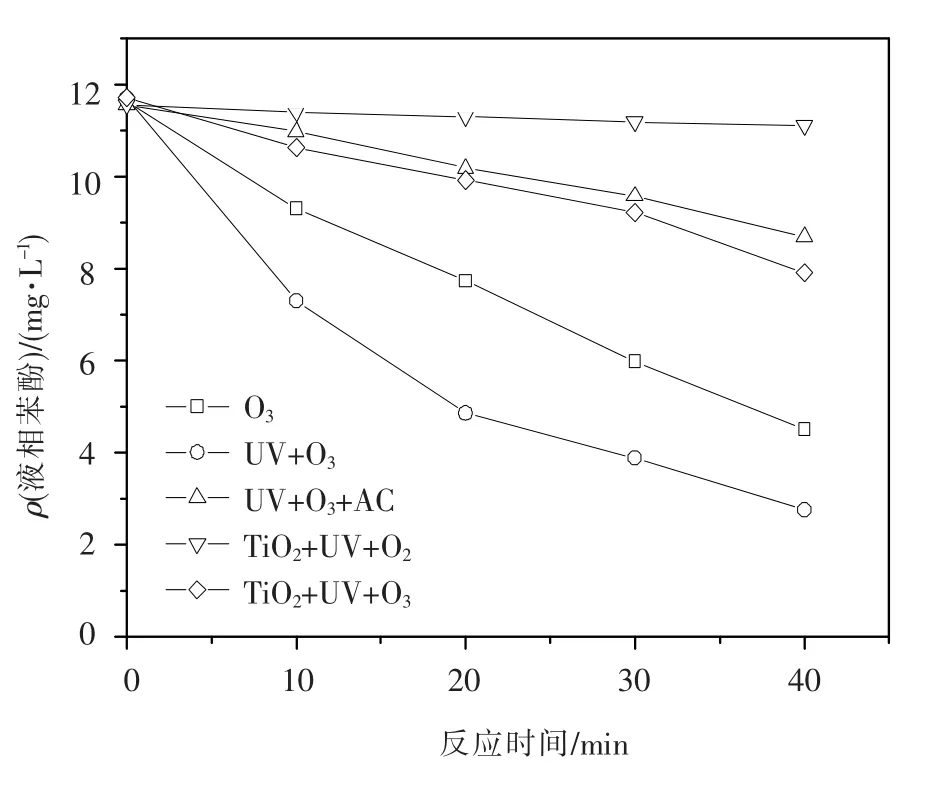
图4 不同氧化过程对液相苯酚降解程度的影响
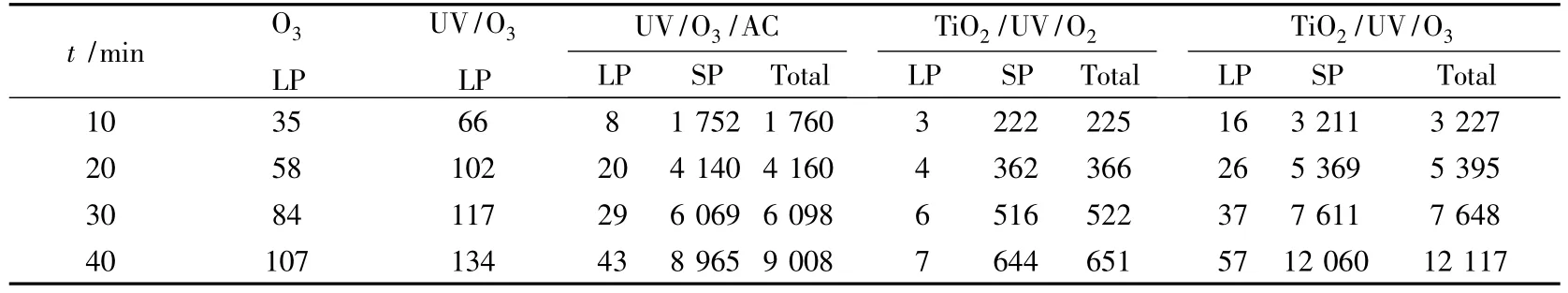
表1 不同氧化过程下苯酚的降解量 mg
2.4 臭氧光催化降解苯酚的动力学模型
由于臭氧与有机物的反应复杂、步骤繁多,在流化床内对其建立数学模型较为困难.为了简化模型,假设所有的总反应均为一级反应.
对于UV/O3/AC系统,液相苯酚质量浓度的改变源自3个过程:液相中的均相反应、AC表面的多相反应以及从AC的吸附/脱附.基于上面的假设,液相中的均相反应动力学方程可表达为
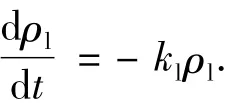
其中kl为均相反应速率常数,可由UV/O3过程的logρ-t图求出.AC表面的多相反应速率与AC表面吸附的苯酚质量浓度ρs以及多相光化臭氧氧化反应速率常数ks成正比,即
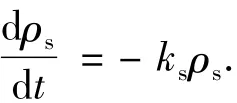
假设吸附到AC表面的苯酚全部参与多相反应,且这部分苯酚在AC表面的体积为Vs,则参与多相反应的量应与吸附的量相等,即
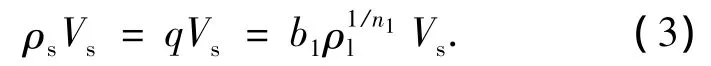
液相质量浓度平衡方程:
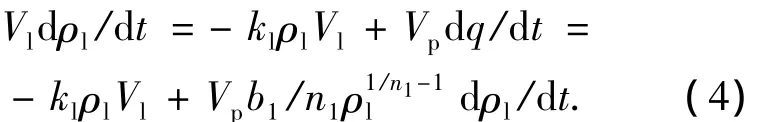
固相表面质量浓度平衡方程:
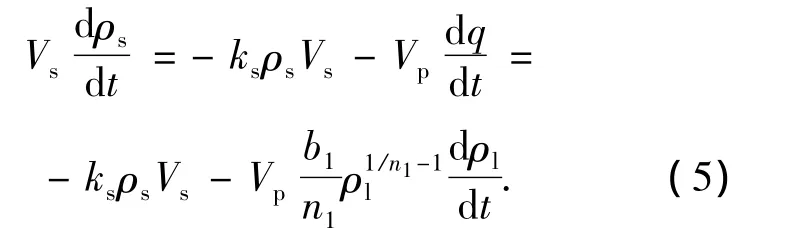
合并方程(3)、(4)、(5)可得
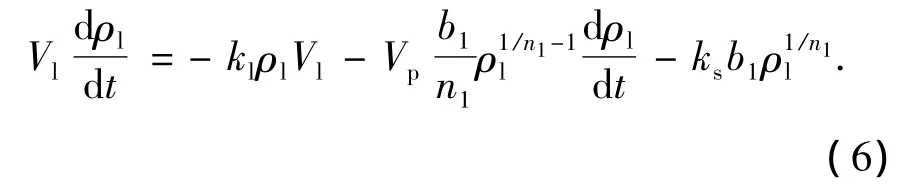
初始条件为
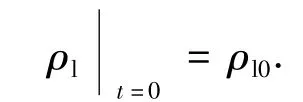
根据初始条件以及UV/O3/AC过程的4个实验数据点,通过数值积分和插值法求出ks,其值与相对标准偏差(DRS)见图5(a).可以看出,本模型与实验数据可以较好地吻合.
对于TiO2-AC/UV/O3过程,其动力学方程应在方程(6)基础上插入光催化反应项,即方程(1),并且吸附/脱附项中AC的特征参数由Ti2-AC替换.对原活性炭与催化剂颗粒的吸附特性研究表明,在稀溶液下两种颗粒对苯酚的吸附/脱附能力相差很小;而且对于流化床中UV/O3/AC和TiO2-AC/UV/O3两个氧化过程其余运行条件均一致,因此ks可以认为不变.采用与UV/O3/AC过程同样的物质平衡法得到苯酚降解动力学方程:
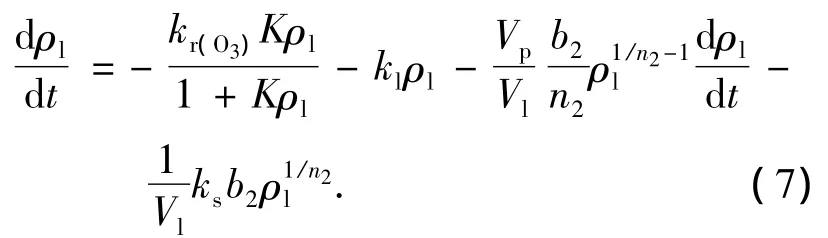
其中kr(O3)为TiO2-AC/UV/O3过程多项光催化反应速率常数.kr(O3)的解法与ks一致,其值与相对标准偏差见图5(b).比较TiO2-AC/UV/O3与TiO2-AC/UV/O2两个过程可以看出,臭氧可以使光催化反应的速率常数提高14.8倍(kr(O3)/ kr=14.8).Sanchez等[20]认为TiO2光催化与臭氧联合作用一般发生如下反应:
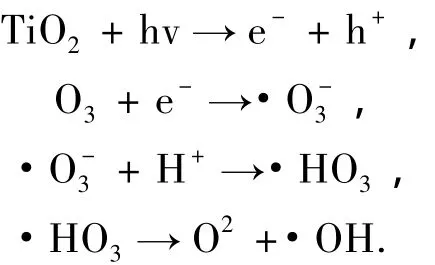
可以看出臭氧捕获光生电子,降低了空穴电子对的复合,同时也生成了更多的羟基自由基,加速了光催化反应的速度.说明在TiO2光催化氧化降解苯酚过程中,臭氧比氧气更适合作为捕获剂.
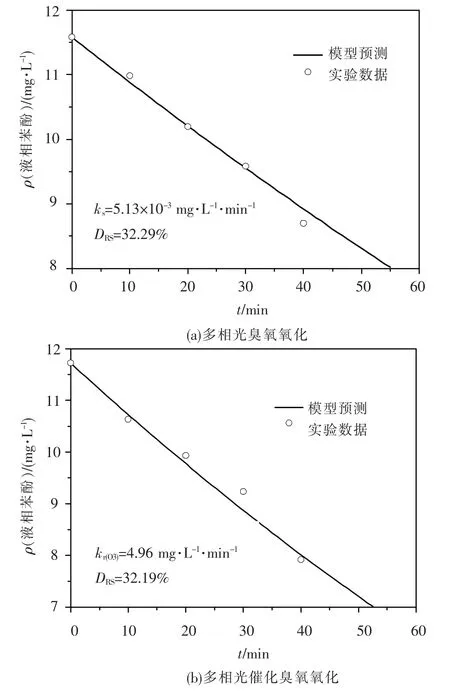
图5 多相光臭氧氧化与多相光催化臭氧氧化系统实验数据与模型预测
3 结论
1)采用溶胶凝胶方法,制备的负载型TiO2-AC光催化剂颗粒,与AC同样在稀溶液下对苯酚的吸附符合Freundlich吸附方程.
2)Langmiur-Hinshelwood动力学方程对流化床中光催化氧化苯酚过程有较好的预测.对于多相反应过程,由于颗粒表面对氧化剂的吸附以及对苯酚的脱附作用,液相中苯酚的降解量小于均相反应.分析认为多相光催化臭氧氧化是均相臭氧氧化、多相光臭氧氧化以及多相光催化的联合,因此对有机物具有较高的去除效率.
3)分别对流化床中UV/O3/AC过程和 TiO2-AC/UV/O3过程建立了简化的反应动力学模型,利用实验数据进行模型参数估值,确定了多相光臭氧氧化反应速率常数以及TiO2-AC/UV/O3过程中多相光催化反应速率常数.模型结果对两种氧化过程中苯酚质量浓度的降低有较好的预测.当以臭氧代替氧气作为捕获剂时,不仅降低了光生空穴电子堆的复合,也产生更多的羟基自由基.模型计算结果表明,多相光催化反应速率常数提高了14.8倍.
[1] CAREY J H,LAWRENCE J,TOSINE H M.Phototdechlorination of PCBs in the presence of titanium dioxide in aqueous suspensions[J].Bull Environ Contam Toxicol,1976,16:697-701.
[2] OLLIS D,PELLIZZETTI E,SERPONE N.Destruction of water contamination[J].Environ Sci Technol,1991,25:1523-1529.
[3] MAZZARINO I,OICCININI P,SPINELLI L.Degradation of organic pollutants in water by photochemical reactors[J].Catal Today,1999,48:315-321.
[4] NAM W S,HAN G Y.A photocatalytic performance of TiO2photocatalyst prepared by the hydrothermal method[J].Korean J Chem Eng,2003,20:180-184.
[5] LASA de H,SERRANO B,SALAICES M.Photocatalytic Reaction Engineering[M].New York:Springer,2005.
[6] KLARE M,WALDNER G,BAUER R,et al.Degradation of nitrogen containing organic compounds by combined photocatalysis and ozonation[J].Chemosphere,1999,38(9):2013-2027.
[7] GIMENO O,RIVAS F J,BELTRAN F J,et al.Photocatalytic ozonation of winery wastewaters[J].J Agric Food Chem,2007,55:9944-9950.
[8] GIRI R R,OZAKI H,TAKANAMI R,et al.Heterogeneous photocatalytic ozonation of 2,4-D in dilute aqueous solution with TiO2fiber[J].Water Science&Technology,2008,58(1):207-216.
[9] SANDRA G M,RENATO S F,NELSON D.Degradation and toxicity reduction of textile effluent by combined photocatalytic and ozonation processes[J]. Chemophere,2000,40:369-373.
[10] LI L S,ZHANG P Y,ZHU W P,et al.Comparison of O3-BAC,UV/O3-BAC and TiO2/UV/O3-BAC process for removing organic pollutants in second effluents[J].Journal of Photochemistry and Photobiology A:Chemistry,2005,171:149-155.
[11] WOLD A.Photocatalytic properties of TiO2[J]. Chem Mater,1993,5:280-283.
[12] HOFFMANN M R,MARTIN S T,CHOI W,et al. Environmental applications of semiconductor photocatalysis[J].Chem Rev,1995,95:69-96.
[13] FUJISHIMA A,RAO T N,TRYK D A.Titanium dioxide photocatalysis[J].J Photochem Photobiol C: Photochem Rev,2000,1:1-21.
[14] MATTHEWS R W,MCEVOY S R.Photocatalytic degradation of phenol in the presence of near-UV illuminated titanium dioxide[J].J Photochem Photobiol A:Chem,1992,64:231-246.
[15] HARRSTRICK A,KUT M O,HEINZLE E.TiO2-assisted degradation of environmentally relevant organic compounds in wastewater using a novel fluidized bed photoreactor[J].Environ Sci Technol,1996,30:817-824.
[16] POZZO R L,GIOMBI J L,BALTANÁS M A,et al. The performance in a fluidized bed reactor of photocatalysts immobilized onto inert supports[J].Catal Today,2000,62:175-187.
[17] KANKI T,HAMASAKI S,SANO N,et al.Water purification in a fluidized bed photocatalytic reactor using TiO2-coated ceramic particles[J].Chem Eng J,2005,108:155-160.
[18] MATSUMURA T,NOSHIROYA D,TOKUMURA M,et al.Simplified model for the hydrodynamics and reaction kinetics in a gas-liquid-solid threephase fluidized-bed photocatalytic reactor:Degradation of o-Cresol with immobilized TiO2[J].Ind Eng Chem Res,2007,46:2637-2647.
[19] NAM W,KIM J,HAN G.Photocatalytic oxidation of methyl orange in a three-phase fluidized bed reactor[J].Chemosphere,2002,47:1019-1024.
[20] SNCHEZ L,PERAL J,DOMÈNECH X.Aniline degradation by combined photocatalysis and ozonation[J].Applied Catalysis B:Environmental,1998,19 (1):59-65.
猜你喜欢
煤气与热力(2021年10期)2021-12-02
昆钢科技(2021年6期)2021-03-09
云南化工(2020年11期)2021-01-14
石油石化绿色低碳(2019年6期)2019-01-14
中学生数理化·八年级物理人教版(2017年12期)2017-04-18
中国蔬菜(2016年8期)2017-01-15
山东工业技术(2016年15期)2016-12-01
合成化学(2015年4期)2016-01-17
少儿科学周刊·少年版(2015年1期)2015-07-07
浙江大学学报(工学版)(2015年1期)2015-03-01
