
Insights into the pyrimidine biosynthetic pathway of human malaria parasite Plasmodium falciparum as chemotherapeutic target
2016-09-12 02:22SudaratanaKrungkraiJerapanKrungkraiUnitofBiochemistryDepartmentofMedicalScienceFacultyofScienceRangsitUniversityPatumthani000ThailandDepartmentofBiochemistryFacultyofMedicineChulalongkornUniversityPathumwanBangkok0330Thailand
Sudaratana R. Krungkrai, Jerapan Krungkrai*Unit of Biochemistry, Department of Medical Science, Faculty of Science, Rangsit University, Patumthani 000, ThailandDepartment of Biochemistry, Faculty of Medicine, Chulalongkorn University, Pathumwan, Bangkok 0330, Thailand
Insights into the pyrimidine biosynthetic pathway of human malaria parasite Plasmodium falciparum as chemotherapeutic target
Sudaratana R. Krungkrai1, Jerapan Krungkrai2*1Unit of Biochemistry, Department of Medical Science, Faculty of Science, Rangsit University, Patumthani 12000, Thailand
2Department of Biochemistry, Faculty of Medicine, Chulalongkorn University, Pathumwan, Bangkok 10330, Thailand
ARTICLE INFO ABSTRACT
Article history:
in revised form 16 March 2016 Accepted 15 April 2016
Available online 20 June 2016
Malaria
Plasmodium falciparum
Pyrimidine biosynthetic pathway Drug target
Drug development
Chemotherapy
Malaria is a major cause of morbidity and mortality in humans. Artemisinins remain as the first-line treatment for Plasmodium falciparum (P. falciparum) malaria although drug resistance has already emerged and spread in Southeast Asia. Thus, to fight this disease, there is an urgent need to develop new antimalarial drugs for malaria chemotherapy. Unlike human host cells, P. falciparum cannot salvage preformed pyrimidine bases or nucleosides from the extracellular environment and relies solely on nucleotides synthesized through the de novo biosynthetic pathway. This review presents significant progress on understanding the de novo pyrimidine pathway and the functional enzymes in the human parasite P. falciparum. Current knowledge in genomics and metabolomics are described, particularly focusing on the parasite purine and pyrimidine nucleotide metabolism. These include gene annotation, characterization and molecular mechanism of the enzymes that are different from the human host pathway. Recent elucidation of the three-dimensional crystal structures and the catalytic reactions of three enzymes: dihydroorotate dehydrogenase, orotate phosphoribosyltransferase, and orotidine 5'-monophosphate decarboxylase, as well as their inhibitors are reviewed in the context of their therapeutic potential against malaria.
1. Introduction
Malaria remains as one of the most deadly diseases in tropical and subtropical endemic countries, with almost half of the world’s populations at risk of infection, estimated at 515 million clinical cases and 1.3 million deaths annually[1-3]. Of the five Plasmodium species that infect humans, including. Plasmodium vivax (P. vivax),Plasmodium malariae, Plasmodium ovale, and Plasmodium knowlesi,Plasmodium falciparum (P. falciparum) is the causative agent of the most lethal and severe form of malaria[1,4,5]. P. vivax, responsible for 25%-40% of the estimated annual cases of malaria worldwide, is seldom fatal but relapses often occur even after a primary infection has cleared[6]. Over the past 50 years, the parasites’ resistance to both chloroquine and sulphadoxine-pyrimethamine has rapidly emerged and is now widespread in the endemic countries[7]. Artemisinin and its derivatives, considered the most rapid acting and efficacious drug, are the first-line drugs for treatment of P. falciparum malaria[8]. However by 2009, resistance to the drug treatment has been reported (Figure 1)[9]. Thus, it is deemed necessary to develop novel antimalarial drugs for malaria chemotherapy[10,11]. Applying lessons learned from malaria research in the post-genomic era, together with increased understanding in genomics, transcriptomics and proteomics[12-16], this review highlights the candidate drug targets for antimalarial drug discovery[11,17-20].

Figure 1. Geographic distribution of antimalarial drug resistant P. falciparum. Data were adapted from the WHO[7,9]. The resistance to two most widely used drugs, chloroquine and sulphadoxine-pyrimethamine, were represented in blue and yellow circles, respectively. The artemisinine resistance,spreading over the mainland of Southeast Asia from Vietnam to Myanmar in 2014, was shown in black dash area.
2. Genomics and metabolomics of malaria parasite
Most of the biochemical knowledge on P. falciparum has focused on the intraerythrocytic life cycle of the parasite owing to over 60 years of research[21-24], as well the established cultivation method for these stages since 1976 by Trager and Jensen[25]. With the availability of complete genome sequences from various Plasmodium species such as, rodent malaria parasites: Plasmodium yoelii[26],Plasmodium berghei (P. berghei), and Plasmodium chabaudi[27];human malaria parasites: P. falciparum[12], Plasmodium knowlesi[5],and P. vivax[6], substantial genome information is now available for comparative analyses. The genomes of the five human Plasmodium species are relatively uniform, ranging from 23 to 27 Mb across 14 chromosomes, and comprising approximately 5 500 genes, in which 51% of these have only one intron. The G+C contents of P. falciparum and P. vivax genomes are 19.4% and 42.3%, respectively. There are two extrachromosomal genomes, the mitochondrion(a linear 6 kb-DNA) and apicoplast (a circular 35 kb-DNA). From genomic, transcriptomic and proteomic data for functional reconstruction, the parasite’s metabolic pathways are now mapped in a public database[28,29]. Analysis of metabolic pathways provides strong conceptual frameworks that allow identification of new drug targets resulting in acceleration of preclinical candidates into the drug pipelines.
Several functional key metabolic pathways responsible for survival of the parasite are identified: anaerobic glycolysis, a short version of tricarboxylic acid cycle (Krebs’ cycle), a simple mitochondrial electron transport system (mtETS), pentose phosphate pathway,apicoplast fatty acid synthesis, phospholipid synthesis, shikimate pathway, glyoxalate pathway, heme biosynthesis, coenzyme A and Q biosynthesis, amino acid metabolism, hemoglobin catabolism,vitamin B1 and B6 synthesis, coenzyme folate biosynthesis,purine and pyrimidine nucleotide synthetic pathways[7,12,28,29]. Pathways, most relevant to the intraerythrocytic stages of the life cycle, have unique properties. Some pathways are operated in two or three different cellular components, e.g., heme biosynthesis compartmentalizes in cytosol, mitochondrion and apicoplast[30]. A number do not exist in the human host cells, e.g., folate biosynthesis[31], which is a target of several antimalarial drugs(pyrimethamine, cycloguanil, sulphadoxine); hemoglobin catabolism for required amino acid precursors as well as release of potentially toxic heme. Other pathways are different from human metabolism,for instance, the malarial type Ⅱ fatty acid synthesis that is similar to the bacterial pathway; and the purine and pyrimidine nucleotide synthetic pathways[28,29].
3. Human and parasite purine and pyrimidine nucleotide metabolisms
Nucleotide metabolism, one of the largest metabolic pathways in human cells, provides the building blocks for DNA and RNA synthesis. The nucleotides are also key players in a wide range of cellular functions, ranging from energy transduction, signaling,syntheses of many biomolecules in carbohydrate and lipid metabolisms[32]. Purine and pyrimidine nucleotides can be provided from de novo biosynthetic pathways or supplied via salvage pathways where nucleobases and nucleosides/deoxynucleosides are recycled from nutrients or from degraded DNA and RNA. In humans, both de novo and salvage pathways are functioning at significant levels for the purine and pyrimidine nucleotide requirements, although the salvage pathways were reported to be more active than the de novo pathways[33,34]. This is true also for bacteria, plant and the free-living nematode Caenorhabditis elegans[35-37].
The pathway itself plays an important role in the activation of the nucleoside-based prodrug forms or analogs that can be used in therapy or in itself serve as a drug target[38]. In rapidly growing cells,including tumor and cancer cells, total cellular purine and pyrimidine nucleotide pools are reportedly imbalance suggesting therapeutic importance[39-41].
The mature erythrocyte, which provides the host environment for P. falciparum growth and multiplication during the intraerythrocytic stages, has a relatively limited ability to salvage purine and has no capacity for de novo purine synthesis. Likewise, the erythrocyte has no ability for de novo synthesis of pyrimidines as inferred from the absence or very low levels for enzymes involved in pyrimidine synthesis. In addition, there is little activity of the salvage pathway for utilization of the pyrimidine bases and nucleosides, uracil, uridine and thymidine, eventhough both purine and pyrimidine nucleosides can be taken up by the host erythrocyte[42].
In the intraerythrocytic stages of P. falciparum, only one of the 10 enzymes for de novo synthesis of purine nucleotide, namely adenylosuccinate lyase which is required to synthesize inosine monophosphate (IMP) from the 5-phosphoribosyl-1-pyrophosphate(PRPP) precursor has been identified[12]. This enzyme also catalyzes the adenosine monophosphate (AMP) synthesis from IMP in the purine salvage pathway. The parasite relies on the salvage of purines or their precursors from the human cell and plasma, particularly for hypoxanthine which is the most abundant purine source in human blood and regarded as the key precursor for other purines[43]. Thus, the great majority of salvaged purine is funneled through hypoxanthine to IMP by a broad substrate specificenzyme, hypoxanthine-guanine-xanthine phosphoribosyltransferase(HGXPRT). However, salvage of purine can also occur via the sequential conversion of adenosine to inosine and hypoxanthine by adenosine deaminase (ADA) and purine nucleoside phosphorylase(PNP), respectively. Adenosine is known to be rapidly imported into the parasite by at least two effective transporters[43-45]. Moreover,ADA is able to use methylthioadenosine from the polyamine biosynthetic pathway to produce methylthioinosine, which is then catalyzed to hypoxanthine by PNP[43-45]. Thus, the parasite ADA functions at the intersection of polyamine metabolism and purine salvage pathway.
In order to obtain adenylate nucleotides, IMP is first catalyzed by adenylosuccinate synthetase to adenylosuccinate (AMPS), which is then converted to (AMP) by adenylosuccinate lyase. To get guanylate nucleotides, IMP dehydrogenase catalyzes the conversion of IMP to xanthosine monophosphate (XMP), which is then converted to guanosine monophosphate (GMP) via a predominant route for GMP production by GMP synthetase. HGXPRT is also able to catalyze, albeit with limited activity, the conversion of xanthine base to XMP and guanine base to GMP[45,46]. The current understanding on purine salvage pathway and interconversion of salvaged purine derivatives to provide ATP, dATP, GTP and dGTP for the synthesis of DNA and RNA nucleic acids, and the linkage of polyamine and purine pathways are summarized in Figure 2. Details of the parasite transporters that allow reutilization of preformed purine nucleosides and bases, the enzymes involved in the purine salvage pathway, including known three-dimensional crystal structures of these enzymes in P. falciparum have been recently reviewed[43,44].
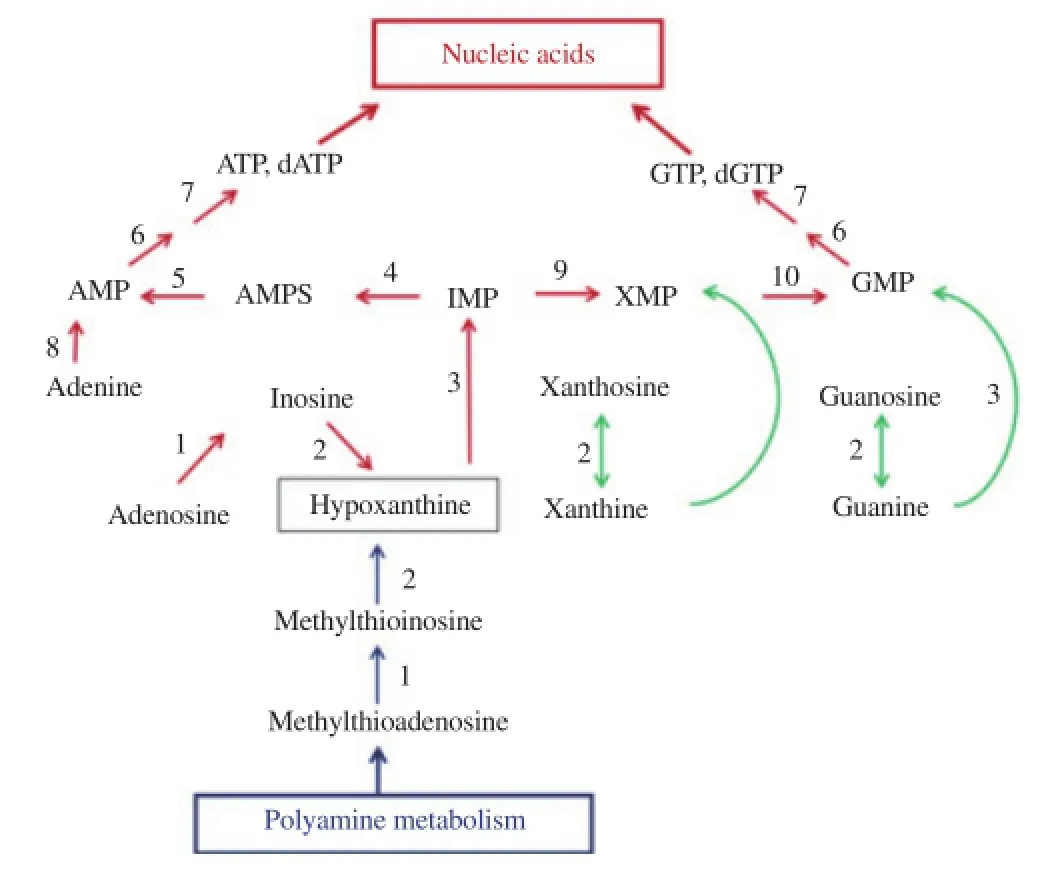
Figure 2. Purine salvage pathway and interconversion of purines in P. falciparum. Red and green arrow indicated predominant and minor pathways,respectively: blue arrow showed polyamine reaction that links purine and polyamine biosynthesis in the parasite. Numbered enzymes were as follows: 1, ADA; 2, PNP; 3, hypoxanthine-guanine-xanthine-phosphoribosyltransferase; 4, adenylosuccinate synthase; 5, adenylosuccinate lyase; 6,nucleoside monophosphate kinase; 7, nucleoside diphosphate kinase;8, adenine phosphoribosyltransferase; 9, IMP dehydrogenase; 10, GMP synthetase. Figure adapted from[42].
4. Malaria parasite de novo pyrimidine biosynthetic pathway
The de novo pyrimidine biosynthetic pathway represents one of the oldest and most conserved metabolic pathway, and the six sequential enzymatic steps starting from bicarbonate (HCO3-), glutamine (Gln),and ATP, producing uridine 5'-monophosphate (UMP) (Figure 3),have remained intact throughout evolution, although the primary structures of the enzymes responsible for the synthesis deviate significantly among prokaryotes, parasitic protozoa, fungi, animals,and mammals including humans[47-51]. As emphasized above, unlike human cells, P. falciparum parasites have little ability to salvage preformed pyrimidine bases and nucleosides from the host cell and plasma, but rely mostly on nucleotide synthesized through the de novo pathway.
Evidence for a functional pyrimidine de novo synthesis in the intraerythrocytic stages comes largely from the following lines of investigation. First, in vitro cultures of P. falciparum incorporate radioactive HCO3-into pyrimidine bases of DNA and RNA. The parasite demonstrates very little incorporation of radioactive orotate, uracil, uridine, thymine and thymidine into its DNA and RNA, whereas the pyrimidines were easily taken up by the P. falciparum-infected erythrocytes[21,22]. Second, although most of the enzymes responsible for salvaging preformed pyrimidine bases and nucleosides have been identified, their activities appears to be low, e.g., thymidine kinase, cytosine kinase, uridine kinase, uracil phosposphoribosyltransferase. There is one exception-the enzyme uridine phosphorylase (UP), required to add ribose from ribose-1-phosphate to uracil for uridine production. The enzyme was identified in both P. falciparum genome (locus: PFE0660c) and the parasite extract[12,52]. The primary structure of the parasite UP suggests that it is one of the moonlighting proteins having both UP and PNP activities on the same polypeptide[53]. Third, all the enzymes required for de novo synthesis of UMP, the first pyrimidine nucleotide metabolite acting as the precursor for synthesis of all pyrimidine nucleotides including CTP, dCTP, TTP, and dTTP, were detected in cell extracts from all Plasmodium species so far examined(Table 1)[54-56]. Finally, the genes encoding each enzymes in all steps of the de novo pathway were detected, whereas most genes for the pyrimidine salvage pathway were conspicuous by their absence with the exception of pfUP gene[12,28,29].
The pyrimidine genes (pyr1 = pfCPSⅡ, pyr2 = pfATC, pyr3 = pfDHO, pyr4 = pfDHOD, pyr5 = pfOPRT, and pyr6 = pfOMPDC)encoding the first six enzymes of the pathway were identified in the P. falciparum genome[12]. The pyr1-pyr6 open reading frames(ORFs) are organized on different locations at various chromosomes: pfCPSⅡ encoding carbamoylphosphate synthetase Ⅱ (CPSⅡ) and pfATC encoding aspartate transcarbamoylase (ATC) on chromosome 13, pfDHO encoding dihydroorotase (DHO) on chromosome 14,pfDHOD encoding dihydroorotate dehydrogenase (DHOD) on chromosome 6, pfOPRT encoding orotate phosphoribosyltransferase(OPRT) on chromosome 5 and pfOMPDC encoding orotidine 5'-monophosphate decarboxylase (OMPDC) on chromosome 10. The location and organization of all six pyrimidine genes are summarizedin Table 2. The parasite pyrimidine genes have the following characteristics: 1) single ORF, having no intron, 2) single copy, and 3) the loci are close to the hypothetical proteins on the chromosome. The genes are separated from each other and is not operon-like in its molecular organization. This property differs from its analogous parasitic protozoan Trypanosoma and Leishmania, where pyr1-pyr6 genes are in an operon-like cluster or synteny constituting of a polycistronic transcript unit on a 25 kb segment of the 800 kb chromosomal DNA[49-50]. The malarial pyrimidine genes are also different from humans in that the fused and single gene pyr1-pyr3-pyr2 (locus: 2p22-21) encodes the multifunctional CAD protein catalyzing the first three enzymes activities and the gene pyr5-pyr6(locus: 3q13) produces the bifunctional UMP synthase activity (Table 3)[47-49]. In addition, the human pyr4 gene is located separately at locus 16q22. In prokaryotes, pyr genes may be localized on different loci as in the bacterial chromosomes, e.g., Escherichia coli (E. coli)[35]; or may be clustered as an operon similar to Bacillus subtilis pyr genes which are transcribed on one large polycistronic message in the 12.5 kb of Bacillus subtilis chromosomal DNA[48].
The identified ORFs of the pyr1- pyr6 genes of P. falciparum are deduced from amino acid sequences of the pyrimidine enzymes. Using multiple sequence alignments and phylogenetic analyses the enzymes pfCPSⅡ, pfDHO and pfOPRT are conserved to bacterial counterparts. The malarial pfATC, pfDHOD and pfOMPDC are mosaic variations, which are homologous to both bacterial and eukaryotic counterparts, including humans. ATC sequence comparison with Toxoplasma gondii, a parasitic protozoan, reveals only 30% identity to the pfATC gene. The pfDHO sequence is close to most bacterial sequences, the yeast Saccharomyces cerevisae and the plant Arabidopsis thalina, indicating that P. falciparum may carry the monofunctional DHO gene acquired thru horizontal gene transfer from a proteobacterium, e.g., E. coli, Neisseria gonorrhoeae[52].
In the malaria parasite, there is relatively little information on the sequential enzymatic steps after UMP synthesis before yielding dCTP, dTTP, CTP, TTP which are the building blocks for DNA and RNA synthesis (Figure 3). Genes are present in the parasite genome but few enzymes have been studied to date. P. falciparum ribonucleotide reductase (RNR) catalyzes the production of deoxyribonucleotides from ribonucleotides, which is associated with thioredoxin reductase[57-59]. TMP kinase (TMPK), catalyzing the synthesis of dTTP and TTP, is known as type Ⅰ enzyme by amino acid sequence but has high efficiency in phosphorylation of 3'-azido-3'-dTMP as well as E. coli type Ⅱ TMPK[60]. Notably, the parasite TMPK can also phospholylate dGMP and dUMP with high specificity, indicating a broad spectrum of substrate specificity[61]. However, CTP synthetase (CTPS) which catalyzes the formation of CTP from UTP, is the only known enzyme for cytosine nucleotide de novo synthesis in the parasite[62-65].
In P. falciparum, the de novo pyrimidine pathway is closely linked to the folate biosynthesis by the thymidylate synthetase (TS) (Figure 3)[66]. The parasite TS is a part of the bifunctional dihydrofolate reductase-thymidylate synthase (DHFR-TS), a validated target for antifolate drugs[67]. The three-dimensional crystal structure of the P. falciparum enzyme and substrate channeling domains have been resolved[68]. It is well recognized that the de novo folate pathway is operating in the parasite, like in bacteria, whereas the human host is incapable of de novo synthesis. In addition, the parasite also has ability to salvage preformed folates from the human host[66,69].

Figure 3. Schematic view of six sequential enzymatic reactions for de novo pyrimidine biosynthesis in P. falciparum.The enzyme CA was proposed to be associated with the pyrimidine pathway. The mtETS was linked to the enzyme DHOD of the pathway, functioning as electron disposal. NMPK, nucleoside monophosphate kinase.

Table 1First six enzymes of de novo pyrimidine biosynthesis in two malaria parasites,P. falciparum, P. berghei, vs host erythrocytes1.

Table 2Organization and character of de novo pyrimidine genes and their enzymes in P. falciparum1.
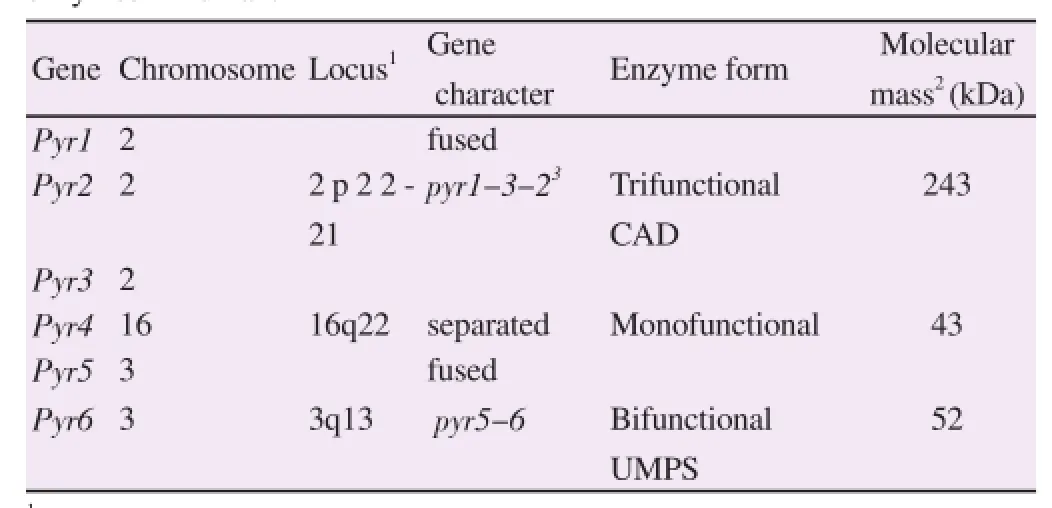
Table 3Organization and molecular property of de novo pyrimidine genes and theirs enzymes in human.
5. Malarial CPSⅡ, ATC and DHO are monofunctional enzymes
Progress towards understanding structures, catalytic mechanisms and regulation of mammalian and human enzymes for de novo pyrimidine pathway has been significant in recent years[32,51]. Certain key differences of the enzymes, pathway, and their genomes including the six enzymes of the pathway from precursors HCO3-,Gln, and ATP to UMP synthesis warrants a closer look.
By using analytical gel filtration chromatography, the first three enzymes (CPSⅡ, ATC, and DHO) of the rodent parasite P. berghei were readily separated into three different molecular masses[70], which is consistent with the presence of three discrete monofunctional proteins. This is similar to that found in another species of protozoa, Crithidia fasciculata, and in many prokaryotic systems[35,70]. The characteristic differs from the humans wherein the CPSⅡ, ATC and DHO activities are carried on a 243-kDa multifunctional protein, called CAD[47].
The malarial DHO enzyme has been purified from P. falciparum and its gene has been cloned, expressed and characterized in detail by our groups[71]. It is a Zn2+enzyme belonging to the amidohydrolase family, sharing characteristics of both mammalian type Ⅰ and eubacterial type Ⅱ DHO by overall amino acid sequence homology,structural properties, kinetics and inhibitor characteristics[71-73].
In mammals, the CPS Ⅱ domain of the CAD is a rate limiting step of the pathway, which is subject to UTP feedback inhibition and allosteric activation by PRPP[39-41]. Responses to allosteric effectors are modulated by phosphorylation, through signaling cascades of mitogen-activated protein kinase (MAP kinase) and protein kinase A (PKA) when demand of the pyrimidine nucleotides is greatest;and by protein kinase C (PKC) when its demand is least[51]. In the bacterial system, the ATC activity is sensitive to feedback inhibition by CTP, and is activated by ATP using allosteric regulation[35]. At present, the malarial CPS Ⅱ and ATC enzymes are still poorly characterized and regulation of these pyrimidine enzymes is largely unknown.
6. Malarial DHOD is the mitochondrial enzyme
Numerous studies have focused on DHOD, the fourth enzyme in the pathway, particularly as a target for antimalarial agents[74-78]. The P. falciparum DHOD gene has been cloned, expressed and characterized[79]. Immunogold labeling localized DHOD in the inner membrane of mitochondrion[77]. The three-dimensional crystal structure of the parasite enzyme has been elucidated and compared to the human DHOD structure[80,81]. Crystal structures of human and parasite DHOD identifies completely different binding sites for the inhibitor leflunomide. The overall structure is αα/β-barrel, similarly to that of other family 2 DHOD of eukaryotic origin (Figure 4A). It contains flavin mononucleotide prosthetic group, ubiquinone binding site and active site for dihydroorotate substrate, consistent with the previous assumptions using kinetic analyses[76,77]. Furthermore, the pyrimidine pathway is linked to the mtETS through the DHOD and ubiquinone coenzyme[77]. The mtETS are valuable targets in malaria chemotherapy[30,82-85].

Figure 4. Three-dimensional structure of three P. falciparum enzymes: DHOD (A), OPRT (B), and OMPDC (C).
All structures were α/β-barrel, withαα-helix shown in violet and β -strand shown in gold. The DHOD, as monomeric form, was in complex with DSM265 triazolpyrimidine inhibitor. OPRT and OMPDC were in dimeric forms as apoenzyme. The DHOD, OPRT, and OMPDC were taken from Protein Data Bank with PDB ID: 5DEL, 4FYM, and 2ZA2, respectively. The models were built using the Java program.
7. Malarial OPRT and OMPDC are multienzyme complex
We characterized the functional, kinetic, and structural properties of OPRT and OMPDC, the fifth and sixth enzymes of the de novo pathway[86-88]. The OPRT and OMPDC enzymes were purified directly from P. falciparum culture. The native enzymes are organized in an αα2β2heterotetramer structure having two subunits each of OPRT and OMPDC[86]. We also expressed both genes in E. coli[87,88]. Co-expression recombinant P. falciparum OPRT and OMPDC genes also exhibited the α2β2complex formation[89,90]. Most recently the parasites’ low complexity region was found to be responsible for the protein-protein interaction in the heterotetrameric formation of the malarial OPRT and OMPDC enzymes, [(OPRT)2(OMPDC)2], as identified by means of a unique insertion of low complexity amino acid sequence characterized by single amino acid repeat which was not seen in homologous proteinsfrom other organisms (Figure 5)[91]. The steady-state kinetics of initial velocity and product inhibition studies of the monofunctional OPRT and OPRT domain in the heterotetrameric complex follows a random sequential kinetic mechanism[87,89].
Furthermore, three-dimensional crystal structures of the parasite OPRT and OMPDC have been elucidated (Figure 4B, 4C)[92-95]and compared to the recently characterized human OPRT and OMPDC structures[32]. Direct decarboxylation mechanism for OMPDC catalysis was proposed based on the crystal structures of the OMPDC-OMP substrate and OMPDC-UMP product complexes. In this catalytic mechanism, the decarboxylation of OMP is initiated by charge repulsion between the C6 carboxylate group of OMP and the carboxyl group at the side chain of Asp136, followed by a proton transfer from Lys138 to the C6 position of UMP, and Lys102 hydrogen bonded to Asp136 instead of the carboxyl group of the pyrimidine ring after decarboxylation. There is a strong network of charged residues around the C6 position that has the potential to destabilize the ground state and stabilize the transition state for the decarboxylation of OMP[93]. By far, the inhibitors of OMPDC have been designed and chemically synthesized for therapeutic potential against malaria using the structure-based drug-design
approach[94,96,97].
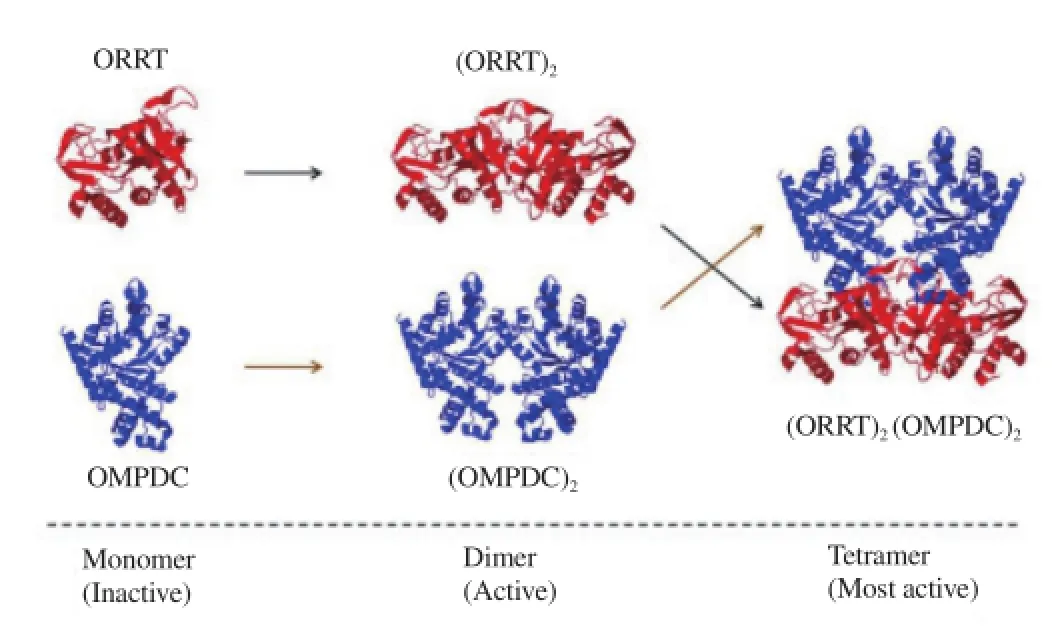
Figure 5. Sequential steps for dimer and tetramer formation of P. falciparum OPRT and OMPDC. The inactive monomer OPRT and OMPDC form its homodimer (OPRT)2and (OMPDC)2. Both homodimers are then associated into heterotetrameric [(OPRT)2(OMPDC)2] complex. The OPRT and OMPDC models are shown in red and blue, respectively.
8. Carbonic anhydrase (CA) is linked to the de novo pyrimidine pathway in the parasite
Functional and kinetic properties of CA were performed from P. falciparum[98]. The parasite CA catalyzes the interconversion of HCO3-and CO2possessing catalytic properties distinct from that of the human host CA isozyme Ⅰ and Ⅱ. Low amino acid sequence identity in the primary structures and phylogenetic analyses is being tapped for malaria chemotherapy, which is now in preclinical phase for drug development[99-101]. The CA supplies HCO3-as substrate for the CPSⅡ of the de novo pyrimidine synthetic pathway, linking the parasite CA to the pyrimidine pathway (Figure 3)[102-104].
9. Concluding remarks and future prospects
Until very recently, artemisinin-resistant parasites have spread over the mainland of Southeast Asian from Vietnam to Myanmar (Figure 1)[105]. The emergence and spread of these parasites entails novel measures for malaria treatment and control. Hopefully, one inhibitor,namely DSM265 (a triazolopyrimidine compound (Figure 4A)),targeting the DHOD enzyme of the pyrimidine pathway would prove promising as it progresses to preclinical and clinical phase Ⅰ trials for drug development[106,107]. Moreover, as structure-based design of antimalarial drug development continues to tapped X-ray crystal structures of the enzyme, in silico screening and surface plasmon resonance analysis, especially for the parasite OMPDC[94], the possibility of modulating potential toxicity through the biochemical pathway might have therapeutic potential against human malaria[108].
Conflict of interest statement
We declare that we have no conflict of interest.
Acknowledgements
We thank Dr. N.M.Q. Palacpac (Biken, Osaka University) for review and grammar correcting of this manuscript. Thanks are also expressed to W. Imprasittichai and P. Paojinda for figure preparation. Our laboratory was supported by the UNDP/World Bank/WHO Special Programme for Research and Training in Tropical Diseases(CHEMAL, TDR/WHO), the National Science and Technology Development Agency of Thailand (NSTDA Career Development Award), the Thailand Research Fund (TRF Basic Research),the Office of Higher Education Commission (OHEC University Staff Development Consortium), Graduate School and Faculty of Medicine, Chulalongkorn University, Thailand.
References
[1] Hay SI, Okiro EA, Gething PW, Patil AP, Tatem AJ, Guerra CA, et al. Estimating the global clinical burden of Plasmodium falciparum malaria in 2007. Plos Medicine 2010; 7(6): e1000290.
[2] Guerin PJ, Olliaro P, Nosten F, Druilhe P, Laxminarayan R, Blinka F, et al. Malaria: current status of control, diagnosis, treatment, and a proposed agenda for research and development. Lancet Infect Dis 2002;2(9): 564-573.
[3] Pink R, Hudson A, Mouries M-A, Bendig M. Opportunities and challenges in antiparasitic drug discovery. Nature Rev Drug Discov 2005;4: 727-740.
[4] White NJ. Plasmodium knowlesi: the fifth human malaria parasite. Clin Infect Dis 2008; 46(2): 172-173.
[5] Pain A, Bohme U, Berry AE, Mungall K, Finn RD, Jackson AP, et al. The genome of the simian and human malaria parasite Plasmodium knowlesi. Nature 2008; 455(7214): 799-803.
[6] Carlton JM, Adams JH, Silva JC, Bidwell SL, Lorenzi H, Caler E, etal. Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature 2008; 455: 757-763.
[7] Ridley RG. Medical need, scientific opportunity and the drive for antimalarial drugs. Nature 2002; 415: 686-693.
[8] White NJ. Qinghaosu (artemisinin): the price of success. Science 2008;320(5874): 330-334.
[9] Dondorp AM, Nosten F, Yi P, Das D, Phyo A, Tarning J, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 2009; 361(5): 455-467.
[10] Fidock DA, Eastman RT, Ward SA, Meshnick SR. Recent highlights in antimalarial drug resistance and chemotherapy. Trends Parasitol 2008;24(12): 537-544.
[11] Olliaro P, Wells TNC. The global portfolio of new antimalarial medicines under development. Clin Pharmacol Ther 2009; 85(6): 584-595.
[12] Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW,et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 2002; 419: 498-511.
[13] Florens L, Washburn MP, Raine JD, Anthony RM, Grainger M, Haynes JD, et al. A proteomic view of the Plasmodium falciparum life cycle. Nature 2002; 419(6906): 520-526.
[14] Le Roch KG, Zhou Y, Blair PL, Grainger M, Moch JK, Haynes JD, et al. Discovery of gene function by expression profiling of the malaria parasite life cycle. Science 2003; 301(5639): 1503-1508.
[15] Aravind L, Iyer LM, Wellems TE, Miller LH. Plasmodium biology: genomic gleanings. Cell 2003; 115(7): 771-785.
[16] Winzeler EA. Malaria research in the post-genomic era. Nature 2008;455(7214): 751-756.
[17] Craft JC. Challenges facing drug development for malaria. Curr Opin Microbio 2008; 11(5): 428-433.
[18] Fatumo S, Plaimas K, Mallm J-P, Schramm G, Adebiyi E, Oswald M,et al. Estimating novel potential drug targets for Plasmodium falciparum by analyzing the metabolic network of knock-out strains in silico. Infect Genet Evol 2009; 9(3): 351-358.
[19] Gamo F-J, Sanz LM, Vidal J, de Cozar C, Alvarez E, Lavandera JL,et al. Thousands of chemical starting points for antimalarial lead identification. Nature 2010; 465: 305-310.
[20] Guiguemde WA, Shelat AA, Bouck D, Duffy S, Crowther GJ, Davis PH,et al. Chemical genetics of Plasmodium falciparum. Nature 2010; 465: 311-315.
[21] Sherman IW. Biochemistry of Plasmodium (malarial parasites). Microbiol Rev 1979; 43(4): 453-493.
[22] Scheibel LW. Plasmodial metabolism and related organellar function during various stages of the life cycle: carbohydrates. In: Wernsdorfer WH, McGregor I, editors. Malaria: principle and practices of malariology. London: Churchill Livingston; 1988. p.199-212.
[23] Sherman IW. Molecular approaches to malaria. Washington DC; ASM Press; 2005. p.1-542.
[24] Pinney JW, Papp B, Hyland C, Wambua L, Westhead DR, McConkey GA. Metabolic reconstruction and analysis for parasite genomes. Trends Parasitol 2007; 23(11): 548-554.
[25] Trager W, Jensen JB. Human malaria parasites in continuous culture. Science 1976; 193: 673-675.
[26] Carlton JM, Angiuoli SV, Suh BD, Kooij TW, Pertea M, Silva JC, et al. Genome sequence and comparative analysis of the model rodent malaria parasite Plasmodium yoelii. Nature 2002; 419(6906): 512-519.
[27] Hall N, Karras M, Raine JD, Carlton JM, Kooij TWA, Berriman M, et al. A comprehensive survey of the Plasmodium life cycle by genomic,transcriptomic, and proteomic analysis. Science 2005; 307(5706): 82-86.[28] Ginsburg H. Progress in in silico functional genomics: the malaria metabolic pathways database. Trends Parasitol 2006; 22(6): 338-340.
[29] Ginsburg H. Caveat emptor: limitations of the automated reconstruction of metabolic pathways in Plasmodium. Trends Parasitol 2009; 25(1): 37-43.
[30] Krungkrai J, Kanchanaphum P, Pongsabut S, Krungkrai SR. Putative metabolic roles of the mitochondria in asexual blood stages and gametocytes of Plasmodium falciparum. Asian Pac J Trop Med 2008;1(1): 31-49.
[31] Krungkrai J, Krungkrai S. Malaria parasite: genomics, biochemistry and drug target for antimalarial development. Chula Med J 2006; 50: 127-142.
[32] Welin M, Nordlund P. Understanding specificity in metabolic pathwaysstructural biology of human nucleotide metabolism. Biochem Biophys Res Commun 2010; 396(1): 157-163.
[33] Weber G. Biochemical strategy of cancer cells and the design of chemotherapy. Cancer Res 1983; 43: 466-492.
[34] Pels Rijcken WR, Overdijk B, Van Den Eijnden DH, Ferwerda W. Pyrimidine nucleotide metabolism in rat hepatocytes: evidence for compartmentation of nucleotide pools. Biochem J 1993; 293(Pt 1): 207-213.
[35] O’Donovan GA, Neuhard J. Pyrimidine metabolism in microorganisms. Bacteriol Rev 1970: 34(3): 278-343.
[36] Katahira R, Ashihara H. Profiles of pyrimidine biosynthesis, salvage and degradation in disks of potato (Solanum tuberosum L.) tubers. Planta 2002; 215(5): 821-828.
[37] Kim S, Park D-H, Kim TH, Hwang M, Shim J. Functional analysis of pyrimidine biosynthesis enzymes using the anticancer drug 5-fluorouracil in Caenorhabditis elegans. FEBS Journal 2009; 276(17): 4715-4726.
[38] Deville-Bonne D, El Amri C, Meyer F, Chen Y, Agrofoglio LA, Janin J. Human and viral nucleoside/nucleotide kinase involved in antiviral drug activation: structural and kinetic properties. Antiviral Res 2010; 86(1): 101-120.
[39] Quemeneur L, Beloeil L, Michallet MC, Angelov G, Tomkowiak M,Revillard JP, Marvel J. Restriction of de novo nucleotide biosynthesis interferes with clonal expansion and differentiation into effector and memory CD8 T cells. J Immunol 2004; 173(8): 4945-4952.
[40] Fairbanks LD, Bofil M, Ruckemann K, Simmonds HA. Importance of ribonucleotide availability to proliferating T-lymphocytes from healthy humans: disproportionate expansion of pyrimidine pools and contrasting effects of de novo synthesis inhibitors. J Biol Chem 1995; 270: 29682-29689.
[41] Weber G. Ordered biochemical program of gene expression in cancer cells. Biochemistry (Moscow) 2001; 66(10): 1164-1173.
[42] Krungkrai J. Dihydroorotase and dihydroorotate dehydrogenase as a target for antimalarial drugs. Drugs Fut 1993; 18: 441-450.
[43] Hyde J. Targeting purine and pyrimidine metabolism in human apicomplexan parasites. Curr Drug Targets 2007; 8(1): 31-47.
[44] Downie MJ, Kirk K, Mamoun CB. Purine salvage pathways in the intraerythrocytic malaria parasite Plasmodium falciparum. Eukaryotic Cell 2008; 7(8): 1231-1237.
[45] Sherman IW. Salvage of purines. Adv Parasitol 2009; 67: 139-145.
[46] Gero AM, O’Sullivan WJ. Purines and pyrimidines in malarial parasites. Blood Cells 1990; 16(2-3): 467-484.
[47] Jones ME. Pyrimidine nucleotide biosynthesis in animals: genes,enzymes, and regulation of UMP biosynthesis. Annu Rev Biochem 1980;49: 253-279.
[48] Quinn CL, Stephenson BT, Switzer RL. Functional organization and nucleotide sequence of the Bacillus subtilis pyrimidine biosynthetic operon. J Biol Chem 1991; 266(14): 9113-9127.
[49] Gao G, Nara T, Shimada JN, Aoki T. Novel organization and sequence of five genes encoding all six enzymes for de novo pyrimidine biosynthesis in Trypanosoma cruzi. J Mol Biol 1999; 285(1): 149-161.
[50] Nara T, Hshimoto T, Aoki T. Evolutionary implications of the mosaic pyrimidine biosynthetic pathway in eukaryotes. Gene 2000; 257(2): 209-222.
[51] Evans DR, Guy HI. Mammalian pyrimidine biosynthesis: fresh insights into an ancient pathway. J Biol Chem 2004; 279: 33035-33038.
[52] Krungkrai J, Prapunwattana P, Wichitkul C, Reungprapavut S,Krungkrai SR, Horii T. Molecular biology and biochemistry of malaria parasite pyrimidine biosynthetic pathway. Southeast Asian J Trop Med Public Health 2003; 34(S2): 32-43.
[53] Kicska GA, Tyler PC, Evans GB, Furneaux RH, Kim K, Schramm VL. Transition state analogue inhibitors of purine nucleoside phosphorylase from Plasmodium falciparum. J Biol Chem 2002; 277: 3219-3225.
[54] Reyes P, Rathod PK, Sanchez DJ, Mrema JE, Rieckmann KH, Ullman B. Enzymes of purine and pyrimidine metabolism from the human malaria parasite, Plasmodium falciparum. Mol Biochem Parasitol 1982;5(5): 275-290.
[55] Gero AM, Tetley K, Coombs GH, Phillips RS. Dihydroorotate dehydrogenase, orotate phosphoribosyltransferase and orotidine 5’-phosphate decarboxylase in Plasmodium falciparum. Trans Roy Soc Trop Med Hyg 1981; 75: 719-720.
[56] Gero AM, Brown GV, O’Sullivan WJ. Pyrimidine de novo synthesis during the life cycle of the intraerythrocytic stage of Plasmodium falciparum. J Parasit 1984; 70(4): 536-541.
[57] Rubin H, Salem JS, Li LS, Yang FD, Mama S, Wang ZM, et al. Cloning,sequence determination, and regulation of the ribonucleotide reductase subunits from Plasmodium falciparum: a target for antimalarial therapy. Proc Natl Acad Sci USA 1993; 90: 9280-9284.
[58] Chakrabarti D, Schuster SM, Chakrabarti R. Cloning and characterization of the subunit genes of ribonucleotide reductase, a cellcycle-regulated enzyme, from Plasmodium falciparum. Proc Natl Acad Sci USA 1993; 90: 12020-12024.
[59] Reichard P. Ribonucleotide reductase: substrate specificity by allostery. Biochem Biophys Res Commun 2010; 396(1): 19-23.
[60] Whittingham JL, Carrero-Lerida J, Brannigan JA, Ruiz-Perez LM, Silva AP, Fogg MJ, et al. Structural basis for the efficient phosphorylation of AZTMP and dGMP by Plasmodium falciparum type Ⅰ thymidylate kinase. Biochem J 2010; 428(3): 499-509.
[61] Kandeel M, Kitamura Y, Kitade Y. The exceptional properties of Plasmodium deoxyguanylate pathways as a potential area for metabolic and drug discovery studies. Nucleic Acids Symp Ser 2009; 53(1): 39-40.
[62] Perignon JL, Hamet M, Druilhe P. Cytidine triphosphate synthetase activity in asexual blood stages of Plasmodium falciparum. Mol Biochem Parasitol 1994; 67: 179-182.
[63] Hendricks EF, O’Sullivan WJ, Stewart TS. A cytidine triphosphate synthetase gene in Plasmodium falciparum. Int J Parasitol 1994; 24: 397-399.
[64] Hendricks EF, O’Sullivan WJ, Stewart TS. Molecular cloning and characterization of the Plasmodium falciparum cytidine triphosphate synthetase gene. Biochim Biophys Acta 1998; 1399(2-3): 213-218.
[65] Yuan P, Hendricks EF, Fernandez HR, O’Sullivan WJ, Stewart TS. Functional expression of the gene encoding cytidine triphosphate synthetase from Plasmodium falciparum which contains two novel sequences that are potential antimalarial targets. Mol Biochem Parasitol 2005; 143(2): 200-208.
[66] Krungkrai J, Webster HK, Yuthavong Y. Folate and cobalamin metabolism in Plasmodium falciparum. Parasitol Today 1990; 6(12): 388-391.
[67] Yuthavong Y, Yuvaniyama J, Chitnumsub P, Vanichtanankul J,Chusacultanachai S, Tarnchompoo B, et al. Malarial (Plasmodium falciparum) dihydrofolate reductase-thymidylate synthase: structural basis for antifolate resistance and development of effective inhibitors. Parasitology 2005; 130(Pt3): 249-259.
[68] Yuvaniyama J, Chitnumsub P, Kamchonwongpaisan S, Vanichtanankul J, Sirawaraporn W, Taylor P, et al. Insights into antifolate reductase from malarial DHFR-TS structures. Nat Struct Biol 2003; 10: 357-365.
[69] Sherman IW. De novo synthesis of pyrimidines and folates. Adv Parasitol 2009; 67: 117-138.
[70] Krungkrai J, Cerami A, Henderson GB. Pyrimidine biosynthesis in parasitic protozoa: purification of a monofunctional dihydroorotase from Plasmodium berghei and Crithidia fasciculata. Biochemistry 1990; 29: 6270-6275.
[71] Krungkrai SR, Wutipraditkul N, Krungkrai J. Dihydroorotase of human malarial parasite Plasmodium falciparum differs from host enzyme. Biochem Biophys Res Commun 2008; 366(3): 821-826.
[72] Porter TN, Li Y, Raushel FM. Mechanism of the dihydroorotase reaction. Biochemistry 2004; 43: 16285-16292.
[73] Lee M, Chan CW, Graham SC, Christopherson RI, Guss JM, Maher MJ. Structures of ligand-free and inhibitor complexes of dihydroorotase from Escherichia coli: implications for loop movement in inhibitor design. J Mol Biol 2007; 370(5): 812-825.
[74] Krungkrai J, Krungkrai SR, Phakanont K. Antimalarial activity of orotate analogs that inhibit dihydroorotase and dihydroorotate dehydrogenase. Biochem Pharmacol 1992; 43(6): 1295-1301.
[75] Seymour KK, Lyons SD, Phillips L, Rieckmann KH, Christopherson RI. Cytotoxic effects of inhibitors of de novo pyrimidine biosynthesis upon Plasmodium falciparum. Biochemistry 1994; 33(17): 5268-5274.
[76] Krungkrai J, Cerami A, Henderson GB. Purification and characterization of dihydroorotate dehydrogenase from the rodent malaria parasite Plasmodium berghei. Biochemistry 1991; 30(7): 1934-1939.
[77] Krungkrai J. Purification, characterization and localization of mitochondrial dihydroorotate dehydrogenase in Plasmodium falciparum,human malaria parasite. Biochim Biophys Acta 1995; 1243(3): 351-360.
[78] Heikkila T, Ramsey C, Davies M, Galtier C, Stead AMW, Johnson AP,et al. Design and synthesis of potent inhibitors of the malaria parasite dihydroorotate dehydrogenase. J Med Chem 2007; 50(2): 186-191.
[79] LeBlanc SB, Wilson CM. The dihydroorotate dehydrogenase gene homologue of Plasmodium falciparum. Mol Biochem Parasitol 1993; 60: 349-352.
[80] Hurt DE, Widom J, Clardy J. Structure of Plasmodium falciparum dihydroorotate dehydrogenase with a bound inhibitor. Acta Cryst 2006;62(Pt 3): 312-323.
[81] Liu S, Neidhardt TH, Grossman T, Ocain T, Clardy J. Structures of human dihydroorotate dehydrogenase in complex with antiproliferative agents. Structure 2000; 8(1): 25-33.
[82] Krungkrai J. Structure and function of mitochondria in human malarial pathogen Plasmodium falciparum. Trends Comp Biochem Physiol 2000; 6: 95-107.
[83] Krungkrai J. The multiple roles of the mitochondrion of the malarial parasite. Parasitology 2004; 129(Pt 5): 511-524.
[84] Phillips MA, Rathod PK. Plasmodium dihydroorotate dehydrogenase: a promising target for novel anti-malaria chemotherapy. Infect Disord Drug Targets 2010; 10(3): 226-239.
[85] Rodrigues T, Lopes F, Moreira R. Inhibitors of the mitochondrial electron transport chain and de novo pyrimidine biosynthesis as antimalarials: the present status. Curr Med Chem 2010; 17(10): 929-956.[86] Krungkrai SR, Prapunwattana P, Horii T, Krungkrai J. Orotate phosphoribosyltransferase and orotidine 5'-monophosphate decarboxylase exist as multienzyme complex in human malaria parasite Plasmodium falciparum. Biochem Biophys Res Commun 2004; 318(4): 1012-1018.
[87] Krungkrai SR, Aoki S, Palacpac NMQ, Sato D, Mitamura T, Krungkrai J, et al. Human malaria parasite orotate phosphoribosyltransferase: functional expression, characterization of kinetic reaction mechanism and inhibition profile. Mol Biochem Parasitol 2004; 134(2): 245-255.
[88] Krungkrai SR, DelFraino BJ, Smiley JA, Prapunwattana P,Mitamura M, Horii T, et al. A novel enzyme complex of orotate phosphoribosyltransferase and orotidine 5'-monophosphate decarboxylase in human malaria parasite Plasmodium falciparum: physical association, kinetics and inhibition characterization. Biochemistry 2005; 44(5): 1643-1652.
[89] Kanchanaphum P, Krungkrai J. Kinetic benefits and thermal stability of orotate phosphoribosyltransferase and orotidine 5'-monophosphate decarboxylase enzyme complex in human malaria parasite Plasmodium falciparum. Biochem Biophys Res Commun 2009; 390(2): 337-341.
[90] Kanchanaphum P, Krungkrai J. Co-expression of human malaria parasite Plasmodium falciparum orotate phosphoribosyltransferase and orotidine 5'-monophosphate decarboxylase as enzyme complex in Escherichia coli: a novel strategy for drug development. Asian Biomed 2010; 4(2): 297-306.
[91] Imprasittichai W, Roytrakul S, Krungkrai SR, Krungkrai J. A unique insertion of low complexity amino acid sequence underlines protein-protein interaction in human malaria parasite orotate phosphoribosyltransferase and orotidine 5'-monophosphate decarboxylase. Asian Pac J Trop Med 2014; 7: 184-192.
[92] Krungkrai SR, Kusakari Y, Tokuoka K, Inoue T, Adachi H, Matsumura H, et al. Crystallization and preliminary crystallographic analysis of orotidine 5'-monophosphate decarboxylase from human malaria parasite Plasmodium falciparum. Acta Cryst 2006; 62(Pt 6): 542-545.
[93] Tokuoka K, Kusakari Y, Krungkrai SR, Matsumura H, Krungkrai J,Horii T, et al. Structural basis for the decarboxylation of orotidine 5'-monophosphate (OMP) by Plasmodium falciparum OMP decarboxylase. J Biochem 2008; 143(1): 69-78.
[94] Takashima Y, Mizohata E, Krungkrai SR, Fukunishi Y, Kinoshita T,Sakata T, et al. The in silico screening and X-ray structure analysis of the inhibitor complex of Plasmodium falciparum OMP decarboxylase. J Biochem 2012; 152(2): 133-138.
[95] Takashima Y, Mizohata E, Tokuoka K, Krungkrai SR, Kusakari Y,Konoshi S, et al. Crystallization and preliminary crystallographic analysis of orotate phosphoribosyltransferase from human malaria parasite Plasmodium falciparum. Acta Cryst 2012; F68: 244-246.
[96] Bello AM, Poduch E, Fujihashi M, Amani M, Li Y, Crandall I, et al. A potent, covalent inhibitor of orotidine 5'-monophosphate decarboxylase with antimalarial activity. J Med Chem 2007; 50(5): 915-921.
[97] Meza-Avina ME, Wei L, Buhendwa MG, Poduch E, Pai EF, Kotra LP. Inhibition of orotidine 5'-monophosphate decarboxylase and its therapeutic potential. Min Rev Med Chem 2008; 8(3): 239-247.
[98] Krungkrai SR, Suraveratum N, Rochanakij S, Krungkrai J. Characterisation of carbonic anhydrase in Plasmodium falciparum. Int J Parasitol 2001; 31(7): 661-668.
[99] Krungkrai J, Scozzafava A, Reungprapavut S, Krungkrai SR, Rattanajak R, Kamchonwongpaisan S, et al. Carbonic anhydrase inhibitors. Inhibition of Plasmodium falciparum carbonic anhydrase with aromatic sulfonamides: towards antimalarials with a novel mechanism of action?Bioorg Med Chem 2005; 13(2): 483-489.
[100] Krungkrai J, Krungkrai SR, Supuran CT. Malarial parasite carbonic anhydrase and its inhibitors. Curr Top Med Chem 2007; 7(9): 909-917.
[101] Krungkrai J, Supuran CT. The alpha-carbonic anhydrase from the malarial parasite and its inhibition. Curr Pharm Design 2008; 14(7): 631-640.
[102] Krungkrai J, Krungkrai SR, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of Plasmodium falciparum carbonic anhydrase with aromatic/ heterocyclic sulfonamides: in vitro and in vivo studies. Bioorg Med Chem Lett 2008; 18(20): 5466-5471.
[103] Krungkrai J, Krungkrai SR, Supuran CT. Malaria parasite carbonic anhydrase and its inhibition in the development of novel therapies of malaria. In: Supuran CT, Winun JY. (eds.) Drug design of zinc-enzyme inhibitors. Hoboken: John Wiley & Sons, Inc; 2009. p. 335-357.
[104] Krungkrai, SR, Krungkrai, J. Malaria parasite carbonic anhydrase: inhibition of aromatic/heterocyclic sulfonamides and its therapeutic potential. Asian Pac J Trop Biomed 2011; 1(3): 233-242.
[105] Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, et al. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 2014; 371(5): 411-423.
[106] Phillips MA, Gujjar R, Malmquist NA, White J, El Mazouni F, Baldwin J, et al. Triazolopyrimidine-based dihydroorotate dehydrogenase inhibitors with potent and selective activity against the malaria parasite Plasmodium falciparum. J Med Chem 2008; 51(12): 3649-3653.
[107] Gujjar R, Marwaha A, El Mazouni F, White J, White KL, Creason S,et al. Identification of a metabolically stable triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with antimalarial activity in mice. J Med Chem 2009; 52(7): 1864-1872.
[108] Flannery EL, Chatterjee AK, Winzeler EA. Antimalarial drug discoveryapproaches and progress towards new medicines. Nature Rev Micro 2013; 11(12): 849-862.
Document heading 10.1016/j.apjtm.2016.04.012
15 February 2016
*
Dr. Jerapan Krungkrai, Department of Biochemistry, Faculty of Medicine, Chulalongkorn University, 1873 Rama 4 Rd., Pathumwan, Bangkok 10330, Thailand.
E-mail: jerapan.k@chula.ac.th; jerapan.k@gmail.com
Fax: (662)-2524986
Tel: (662) 2564482
 Asian Pacific Journal of Tropical Medicine2016年6期
Asian Pacific Journal of Tropical Medicine2016年6期
- Asian Pacific Journal of Tropical Medicine的其它文章
- Experiment research of cisplatin implants inhibiting transplantation tumor growth and regulating the expression of KLK7 and E-cad of tumor-bearing mice with gastric cancer
- Correlation study of biological characteristics of non-small cell lung cancer A549 cells after transfecting plasmid by microbubble ultrasound contrast agent
- Expression and significance of angiostatin, vascular endothelial growth factor and matrix metalloproteinase-9 in brain tissue of diabetic rats with ischemia reperfusion
- Change of the peripheral blood immune pattern and its correlation with prognosis in patients with liver cancer treated by sorafenib
- Comparative study of the chitooligosaccharides effect on the proliferation inhibition and radiosensitization of three types of human gastric cancer cell line
- Are efforts up to the mark? A cirrhotic state and knowledge about HCV prevalence in general population of Pakistan