
基于多重相似性和增强注意力预测药物-靶标相互作用
2025-03-04 00:00:00王伟余梦雪孙斌万仕彤刘栋周运张红军王鲜芳
河南师范大学学报(自然科学版) 2025年2期
关键词:药物






摘 要:在新药发现和药物重定位研究中,发现药物与靶标之间的相互作用是重要的研究内容.针对药物与靶标相互作用网络,提出一种基于多重相似性和增强注意力机制的图卷积神经网络模型(RSGCN)预测药物-靶标相互作用.首先,提出了多重相似性来捕捉网络结构特征,以充分利用节点间的直接或间接关系.然后,通过PCA降维减少相似性噪声对实验结果的影响.最后,采用图卷积神经网络(graph convolution neural network,GCN)获得节点嵌入表示,并融入基于注意力的增强层,通过增强注意力机制获得节点间的注意力权重,能够高效地预测药物与靶标之间的相互作用.在黄金标准数据集上的实验结果表明RSGCN模型具有较好的性能.
关键词:图卷积神经网络(GCN);多重相似性;PCA;增强注意力机制;药物-靶标相互作用
中图分类号:TP181""""" 文献标志码:A文章编号:1000-2367(2025)02-0099-09
药物重定位(或称为药物再利用)是一种对已批准药物发现其新用途的重要策略,为药物研发提供了一种快速高效的新途径.药物研发是一个系统工程,其中药物-靶标的发现和验证是重要的研发过程之一,确定药物-靶标相互作用(DTI)是药物开发和药物重新定位早期阶段的关键步骤[1-2].然而,由于采用湿实验方法鉴定药物-靶标相互作用(DTI)存在研发费用高且研发周期长的问题,因此,采用计算方法从大量候选药物中筛选潜在的DTI,能够减轻费用昂贵和耗时的湿实验研究工作,提高药物发现的效率.
近年来,图神经网络的快速发展将深度学习应用扩展到了图领域,并已应用于基于网络的药物发现[3].基于网络的方法通常包括两个步骤:网络构建和DTI预测.这些方法不仅考虑药物之间的关系,而且考虑靶点之间的关系,因此基于网络的方法受到越来越多的关注[4],通过网络嵌入整合药物和靶点的各种信息,可以进一步提高DTI预测的准确性[5-7].一些研究工作在此领域取得了重要进展.例如,SHANG等[8]提出了一种基于多视图网络嵌入方法来预测潜在的DTI,整合了药物和靶标的异质信息.MHADTI[9]是一种基于多视角异构信息网络的药物-靶点相互作用预测,利用药物和靶点的多源信息构建不同的相似性网络.DTI-HETA[10]利用图卷积神经网络获得药物和靶标的嵌入表示.为了突出不同邻域节点对中心节点聚集图卷积信息的贡献,在节点嵌入过程中引入了图注意机制.KronRLS-MKL[11]能够集成多个异构信息源,适用于任意不同的网络规模,此外,它通过返回权重来自动选择更相关的内核.RTHNEDT[12]是一种基于关系拓扑的网络嵌入方法来预测药物与靶标的相互作用,该模型在带有标签的网络和未带标签的网络都能获得较好的预测性能.HNEDTI[13]模型对药物相似度矩阵和靶标相似度矩阵分别设置两个相似度阈值参数,过滤相似度较低的边,然后用已知的药物相关网络和靶标相关网络构建药物与靶标异质网络.GMDTI[14]是一种基于异
收稿日期:2023-06-28;修回日期:2023-08-03.
基金项目:国家自然科学基金(62072160;62072157);河南省科技攻关项目(242102211045;242102210001).
作者简介(通信作者):王伟(1975-),男,河南新乡人,河南师范大学副教授,博士,研究方向为机器学习、生物信息学和数据挖掘,E-mail:weiwang@htu.edu.cn.
引用本文:王伟,余梦雪,孙斌,等.基于多重相似性和增强注意力预测药物-靶标相互作用[J].河南师范大学学报(自然科学版),2025,53(2):99-107.(Wang Wei,Yu Mengxue,Sun Bin,et al.Prediction of drug-target interaction based on multiple similarity and enhanced attention mechanisms on graph convolution neural network[J].Journal of Henan Normal University(Natural Science Edition),2025,53(2):99-107.DOI:10.16366/j.cnki.1000-2367.2023.06.28.0003.)
质信息并融合元路径信息的图神经网络模型,用于预测药物-靶标相互作用.PDML[15]是一种基于弱标记和多信息融合的药物-靶标相互作用方法.
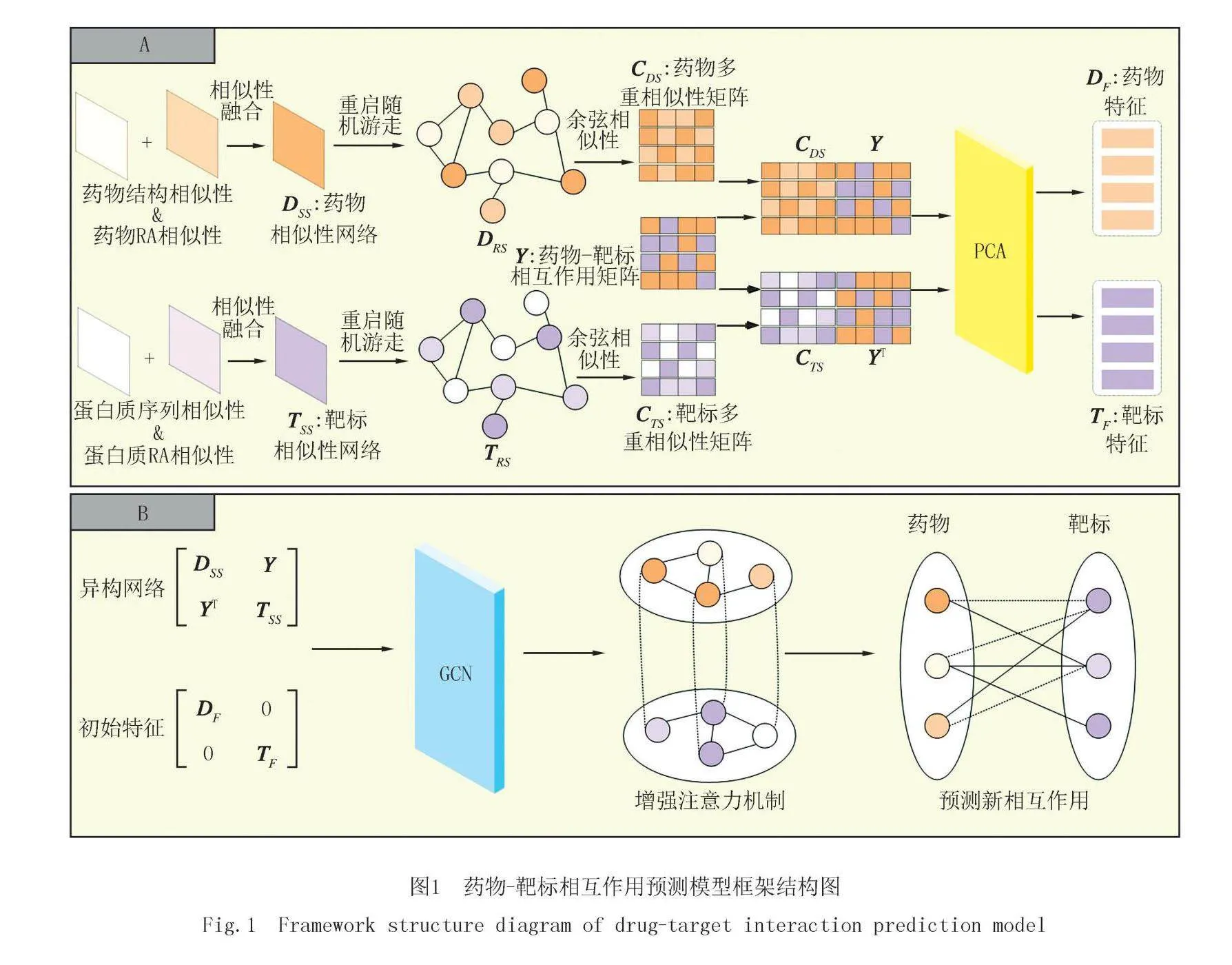
在本研究中,提出了一种基于多重相似性和增强注意力机制的图卷积神经网络模型预测药物与靶标相互作用(RSGCN).与现有的药物-靶标相互作用预测方法不同,模型使用多重相似性模块优化药物和靶标节点的原始特征向量,捕获网络结构特征,以充分获取节点之间的直接或间接关系.具体而言,模型采用相似性融合的方法,获得药物和靶标的相似性网络.通过重启随机游走和余弦相似性方法获得药物和靶标的多重相似性网络.然后,模型使用PCA来降低维度,以减少相似性噪声对计算结果的影响.接下来,通过图卷积神经网络(GCN)提取网络节点特征,以获得节点的低维表示.为了更有效地发现相邻节点之间的关系,模型引入增强注意力机制来获取节点之间的注意力,最终预测药物与靶标之间的相互作用,实验结果与现有的药物-靶标相互作用预测方法相比,RSGCN方法具有良好的性能.模型如图1所示.
1 方 法
1.1 问题定义
将药物和靶标表示为网络中两种不同类型的节点.药物的节点集定义为D={d1,d2,…,dNd}.类似地,靶标节点集定义为T={t1,t2,…,tNt}.其中,Nd表示药物数量,Nt表示靶标数量.网络中的边是药物和靶标之间的关联,可以表示为邻接矩阵Y∈RNd×Nt.矩阵Y是一个只有0和1的矩阵,这里,Yi,j=1指一种药物与一种靶标相互作用.相反,Yi,j=0表示潜在关联或者无关联.本文的任务是获得一个药物-靶标得分矩阵F*.得分越高相互作用的可能性越大,根据得分预测出潜在的药物-靶向相互作用可为药物发现实验提供实验指导,以提高药物研发效率.
1.2 相似性融合
相似性融合可以获得足够的节点信息,有助于提高模型的预测能力.药物相似性网络通过融合药物结构相似性矩阵和药物R相似性(resource allocation similarity)矩阵构建.DSS=d×DS+(1-d)×Rd,(1)
DSS∈RNd×Nd,Nd表示药物数量.d是药物结构相似性矩阵权重系数,DS是药物结构相似性矩阵,DS∈RNd×Nd,实验采用SIMCOMP[16]计算化合物di和dj之间的化学结构相似性,公式如下所示:DS(di,dj)=|di∩dj|/|di∪dj|,(2)
|di∩dj|表示两种化合物di和dj之间的共同子结构的大小,|di∪dj|表示两种化合物di和dj具有子结构的大小.
Rd是R相似性矩阵,Rd∈RNd×Nd.R相似性是对已知的相互作用关系中的邻居节点信息来计算相似性得分.首先为网络中的每个节点分配一个初始单元,然后依据网络中节点间的拓扑关系,把初始单元均匀分发给相邻节点.初始节点与目标节点之间路径上分配的资源总和就是两节点之间的R相似性.例如,节点di和dj节点之间R相似性公式如下:
R(di,dj)=∑r=N(di)∩N(dj)1|N(r)|,(3)
R(di,dj)的值越大,表示起始节点di与目标节点dj之间的相似性越高.其中N(di)和N(dj)分别表示节点di和节点dj的邻居节点集,r和N(di)∩N(dj)表示节点di和节点dj共有的节点集,|N(r)|表示r的邻居节点个数.
靶标相似性网络TSS是通过融合蛋白质序列相似性PS和蛋白质R相似性计算得到,其计算公式如下:TSS=t×PS+(1-t)×Rt,(4)
TSS∈RNt×Nt,Nt表示靶标数量.t是蛋白质序列相似性矩阵权重系数,PS是蛋白质序列相似性矩阵,实验使用标准化版本的Smith-Waterman得分[17]计算蛋白质之间的序列相似性.两种蛋白质ti和tj之间的Smith-Waterman得分如下:PS(ti,tj)=SW(ti,tj)/SW(ti,ti)SW(tj,tj).(5)
类似药物相似性融合,这里Rt指靶标R相似性矩阵,节点ti和节点tj之间R相似性如公式所示:
R(ti,tj)=∑r=N(ti)∩N(tj)1|N(r)|.(6)
1.3 重启随机游走(RWR)
本模型通过RWR来获取节点间的相似性.靶标相似性网络TSS和药物相似性网络DSS通过RWR算法得到靶标特征矩阵TRS和药物特征矩阵DRS.DRS∈RNd×Nd,TRS∈RNt×Nt.RWR[18]的方法定义为:rki=cWrk-1i+(1-c)ei,(7)
其中W=[i,j]是归一化后转移概率矩阵,ei∈Rn+1是第i个节点初始向量(TSS或DSS的行向量),c是重启概率,rki是RWR计算后得到的相似度向量.
1.4 余弦相似性
为充分利用节点之间的关系,模型采用余弦相似性计算方法,公式如下:sim(n,m)=n·m/‖n‖×‖m‖=∑Ni=1(ni×mi)/∑Ni=1n2i×∑Ni=1m2i,(8)
这里ni和mi分别表示节点n和节点m的分量.经过RWR得到的靶标矩阵TRS和药物矩阵DRS,利用余弦相似性得出靶标多重相似性矩阵CTS和药物多重相似性矩阵CDS,其中CDS∈RNd×Nd,CTS∈RNt×Nt.
1.5 特征矩阵融合
为了更好地聚集网络节点信息,模型将靶标多重相似性矩阵CTS和药物多重相似性矩阵CDS分别与药物靶标相互作用矩阵Y相融合,获得靶标特征矩阵ST和药物特征矩阵SD,公式如下:
SD=[CDS Y],(9)
ST=[CTS YT],(10)
其中,SD∈RNd×(Nd+Nt),ST∈RNt×(Nd+Nt).Y∈RNd×Nt,YT∈RNt×Nd.Nd表示药物数量,Nt表示靶标数量.
1.6 主成分分析(PCA)
模型采用PCA降噪和特征提取,经过多重相似性和邻接矩阵Y相结合,噪声往往是不可避免的.为了减少相似性噪声对计算结果的影响,引入了PCA算法.PCA算法能够消除冗余特征,有助于提高处理数据的速度,提升算法的效率.如下所示.
对药物SD矩阵去平均值(即去中心化),即每一位特征减去各自的平均值.计算去中心化矩阵的协方差矩SDD:
SDD=1Nd+NtSDTSD,(11)
其中SDD∈R((Nd+Nt)×(Nd+Nt)).
模型用特征值分解方法求协方差矩阵SDD的特征值与特征向量:
SDD=Q∑Q-1,(12)
其中,Q是矩阵SDD的特征向量矩阵,∑是一个对角矩阵,其对角线上的元素即特征值.
对特征值从大到小排序,选择其中最大的l个,l=(Nd+Nt)×k.然后将其对应的l个特征向量分别作为列向量组成特征向量矩阵Pd.获得去噪后的特征矩阵DF:DF=SD×Pd.(13)
同理,靶标得到特征矩阵TF,TF∈R(Nt×l),DF∈R(Nd×l).
1.7 图卷积神经网络(GCN)
模型通过GCN来获得节点的嵌入表示.为了在降噪后的矩阵中使GCN更容易找到原始节点之间关联,获得更多节点信息,模型中引入了异构网络.在模型中将药物相似性矩阵DSS、靶标相似性矩阵TSS和药物-靶标相互作用矩阵Y构建成异构网络,由邻接矩阵Adt表示,其构建方法如下:Adt=DSSYYTTSS,(14)
这里Adt∈R((Nd+Nt)×(Nd+Nt)).
对于初始特征,构建方法如下:DT=DF00TF,(15)
其中DT∈R((Nt+Nd)×2l).
邻接矩阵Adt与初始特征DT的图卷积计算公式如下:F=(I+D-12AdtD-12)DTWe,(16)
其中,F∈R((Nd+Nt)×LFN)矩阵We表示训练权重矩阵,We∈R(2l×LFN),I表示单位矩阵,D表示Adt的对角矩阵;通过激活函数将偏差矩阵B引入到矩阵F中,得到特征矩阵H:H=ReLU(F+B),(17)
其中H∈R((Nd+Nt)×LFN),B∈R((Nd+Nt)×LFN).
1.8 增强注意力机制
为了使相似的药物(或靶标)节点在特征空间中更好地邻居聚合.本模型引入了增强注意力机制[19],通过学习邻居的权重实现其加权聚合,用于测量邻居节点对节点的影响.aij表示节点之间的注意力系数,如下所示:eij=ReLU(Whi+Whj),(18)
aij=exp(eij)/∑j∈Niexp(eij),(19)
Hi=∑j∈Niaijhi,(20)
其中,Ni表示节点i的邻居节点集,eij表示节点j的特征对节点i的重要性,W表示权重矩阵,ReLU表示激活函数.
1.9 损失函数
模型根据药物和靶标的数量,将特征矩阵H分割成表示药物的特征矩阵Hd和靶标矩阵Ht,计算出药物靶标得分矩阵F*:F*=HdWdHTt,(21)
其中F*∈R(Nd×Nt),Wd是可训练矩阵且Wd∈R(LFN×LFN).
Wnp用于使迭代过程中的预测误差最小化,L为损失函数[18]: L=Wnp+12‖We‖2+12‖Wd‖2+12‖B‖2,(22)
Wnp=∑ij;Φp,ij=1 or Φn,ij=1(M′ij-Mij)/∑ij(Φp,ij+Φn,ij),(23)
其中,Φp和Φn分别代表随机挑选的正负样本矩阵.在Φp中若某个位置的值为1,就代表此元素为正样本.而在Φn中某个位置的值为1,就代表此元素为负样本.M′ij代表模型预测标签,Mij为真实标签.
1.10 RSGCN 算法
RSGCN是一种基于多重相似性和增强注意力机制的图卷积神经网络模型来预测药物与靶标相互作用,其伪代码如表1所示.
1.11 实验数据
本研究采用Yamanishi构建的黄金标准数据集,该数据集药物与靶标主要来自DrugBank[20]、KEGG BRITE[21]、BRENDA[22]和SuperTarget[23]数据库.数据集包括靶向酶(Es)、离子通道(IC)、G蛋白偶联受体(GPCR)和核受体(NRs),共4类数据,其中酶(Es)数据集中药物445个,靶标664个,相互作用对2 926个;离子通道(IC)数据集中药物210个,靶标204个,相互作用对1 476个;G蛋白偶联受体(GPCR)数据集中药物223个,靶标95个,相互作用对635个;核受体(NRs)数据集中药物54个,靶标26个,相互作用对90个.
2 结果与讨论
2.1 参数设置
模型中的学习率设置为0.01.模型采用了学习率自适应的优化算法Adam.潜在因字数LFN(latent_factor_num)表示GCN嵌入维度,在实验时设置为5~100,步长为5,其中LFN为70时模型性能最高;epoch是学习算法在整个训练数据集中的迭代次数,在实验时分别设置为500~2 000,步长为100,在epoch设置为1 000时模型性能最高;dropout分别设置为0.1~0.9,步长为0.1,模型在dropout为0.9时最优;GCN设置1层;在Es数据集上进行了以下消融及对照实验.模型采用10折交叉验证来评估预测方法的性能.
2.2 相似性融合
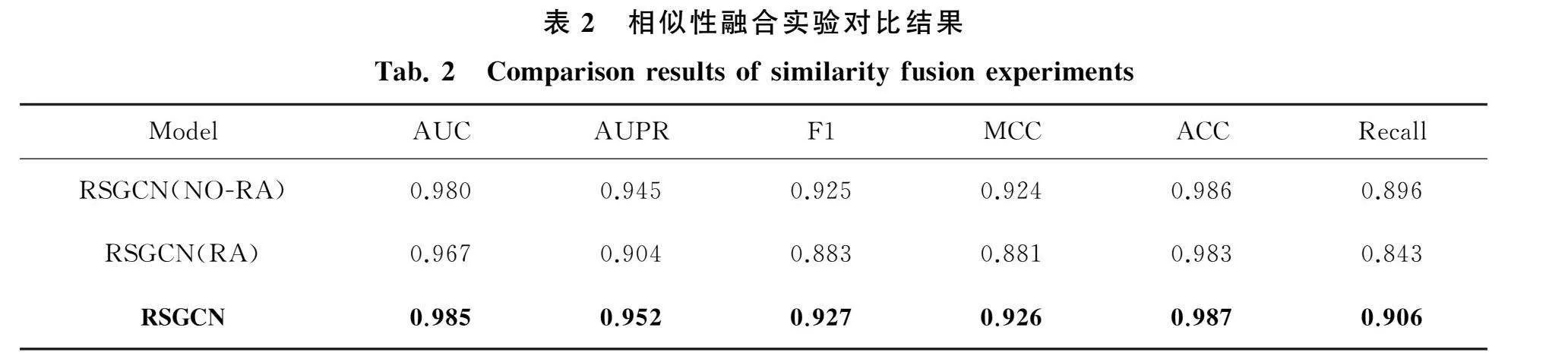
为了验证相似性融合对模型的影响,设置了一组消融实验,如表2所示,RSGCN(NO-RA)表示采用了药物结构相似性和蛋白质序列相似性;RSGCN(R)表示采用了药物R相似性和蛋白质R相似性;RSGCN表示本模型使用了相似性融合.表2明显看出在各个评价指标均高于未使用相似性融合,从而说明相似性融合有助于提高模型的预测能力.
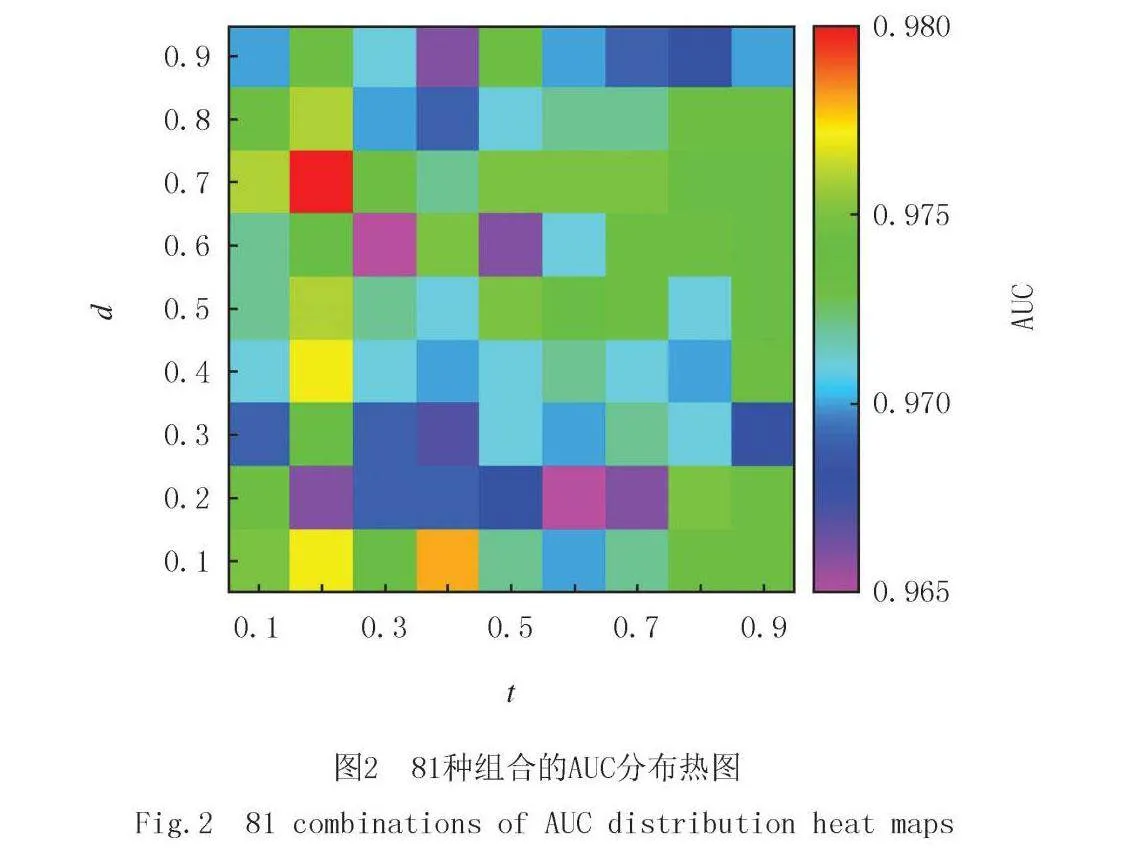
相似性融合是将药物和靶标的两种相似性矩阵融合成一个网络,对药物和靶标融合时有两个重要的权重系数t和d,其中t表示蛋白质序列相似性网络的权重,d表示药物结构相似性网络的权重.为了得到最优权重系数,将t和d分别设置为0.1~0.9,在IC数据集上得到实验结果的AUC如图2所示,t取0.2,d取0.7时AUC达到最高,因此实验中t取0.2,d取0.7.并在其他数据集中也筛选出最优的t和d值,在数据集NRs最优t=0.1,d=0.2;在数据集GPCR最优t=0.6,d=0.4,在数据集Es最优t= 0.9,d=0.3.
2.3 多重相似性
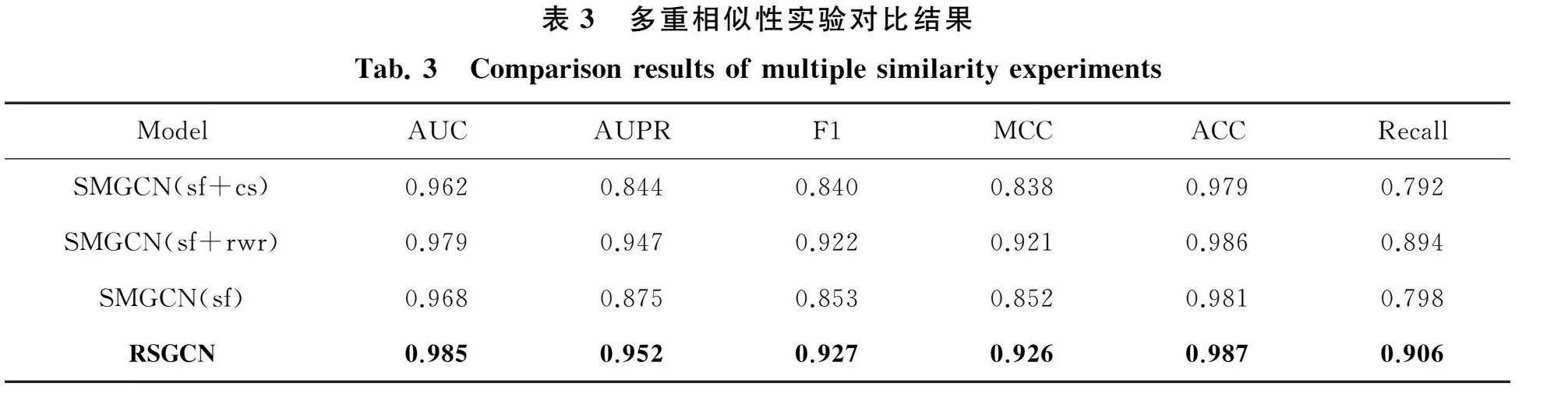
为了验证多重相似性的作用,本文进行了消融实验,如表3所示,SMGCN(sf+cs)表示使用了相似性融合和余弦相似性;SMGCN(sf+rwr)表示使用了相似性融合和RWR;SMGCN(sf)表示只使用了相似性融合;RSGCN是最终的模型,其各项评价指标均达到最高,这是因为多重相似性可以获得更多的节点信息.
2.4 主成分分析(PCA)
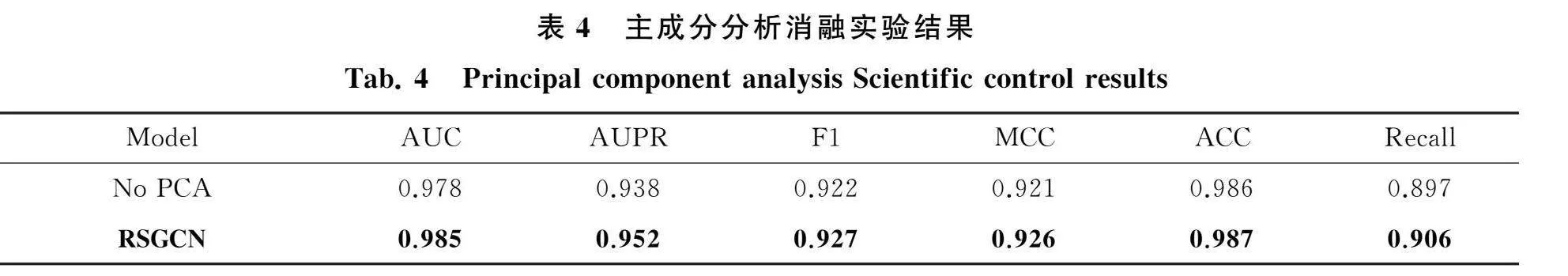
实验进行了使用PCA前后的消融实验.如表4所示,模型在所有评价指标上均优于没有使用PCA,这是因为PCA有助于消除噪声,获取更加显著的特征.
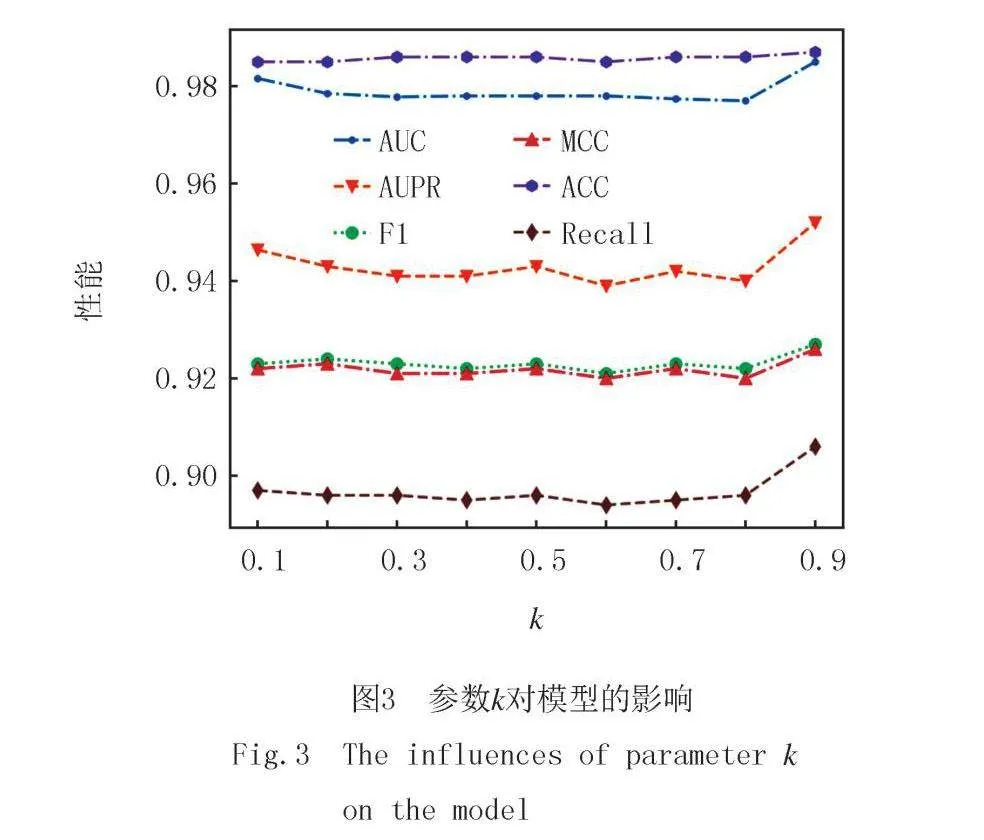
PCA中的参数k,表示维度降为原来的矩阵列数目的k倍.将k取值范围设置为0.1~0.9.如图3所示,当k取0.9时,达到了最优实验性能.
2.5 增强注意力机制
为了使相似的药物(或靶标)节点在特征空间中更好地与邻居聚合.通过增强注意力机制获得节点间的注意力权重.
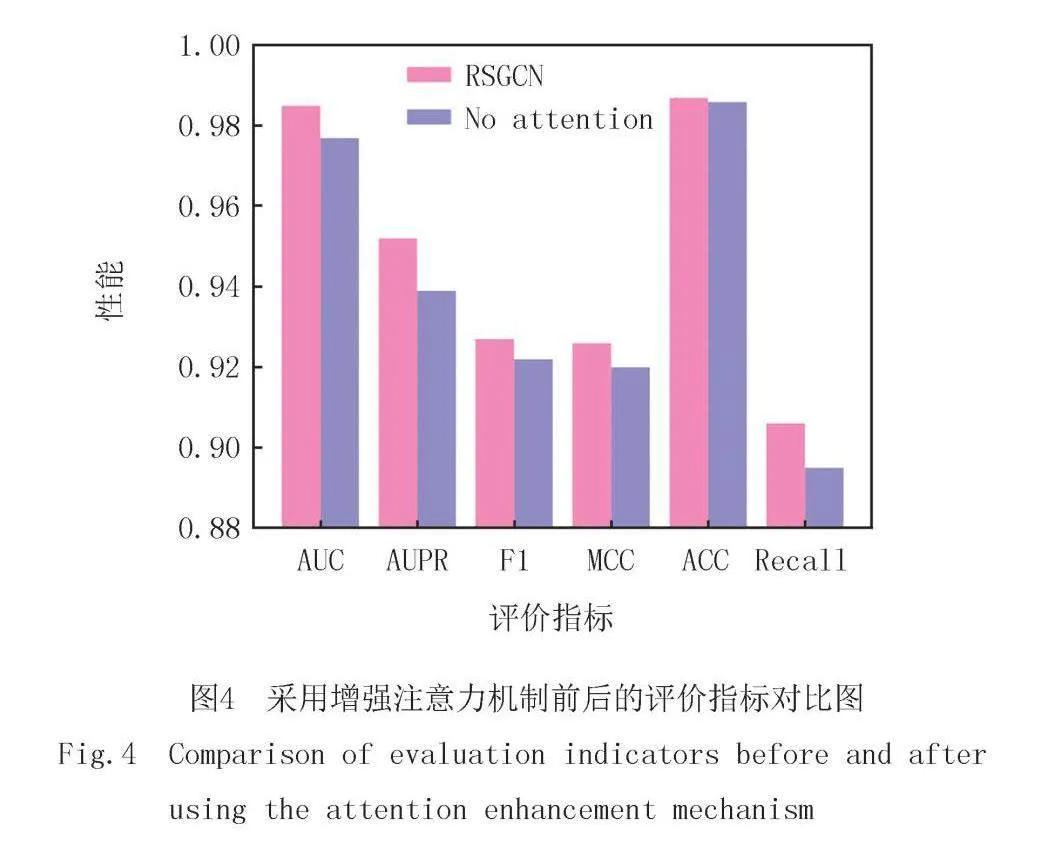
为了验证增强注意力机制的作用,同样设置了一组对照实验,如图4所示,模型在各种评价指标上均优于未使用增强注意力机制的结果,这是因为增强注意力机制能够获得更多邻居节点与节点间的影响.
2.6 与其他方法的比较
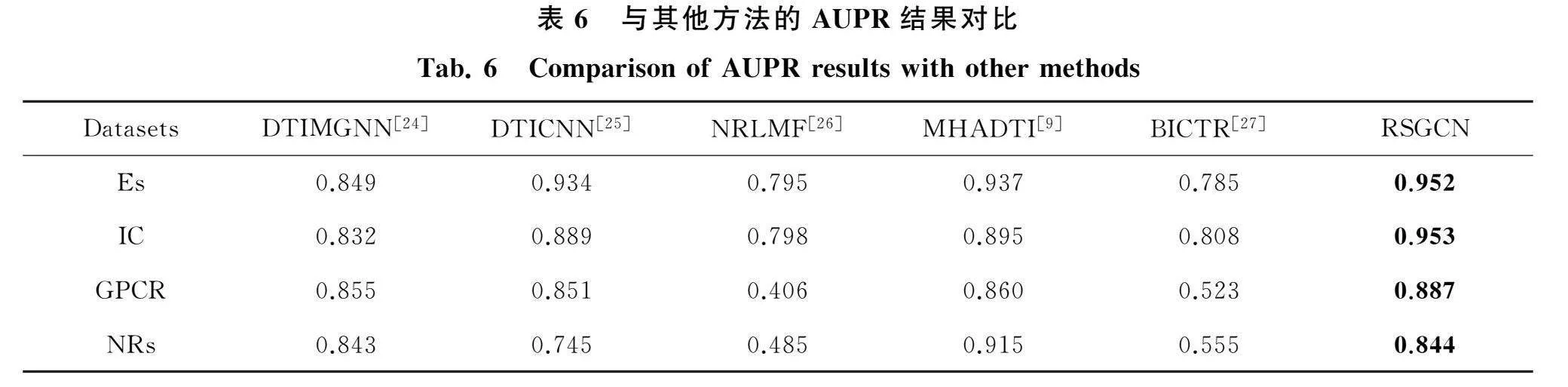
本实验与DTIMGNN[24]、DTICNN[25]、NRLMF[26]、MHADTI[9]和BICTR[27]模型进行了比较.对比结果的AUC和AUPR如表5和表6所示,在数据集Es、IC和NRs上,本模型的性能均优于其他方法.在数据集GPCR,本模型的性能优于绝大多数其他方法.在数据集Es中的AUC分别比DTIMGNN 高7.2%、DTICNN高5.2%、NRLMF高4.5%和MHADTI高4.1%.通过上述分析表明,模型在使用了多重相似性和增强注意力机制后,能够更好地捕捉网络节点信息,充分利用节点间直接或间接的特征,实现了更好的预测性能.
3 总 结
本文提出了一种新颖的基于多重相似性和增强注意力机制的图卷积神经网络模型预测药物与靶标相互作用(RSGCN).首先构建多重相似性矩阵,将药物相似性矩阵和靶标相似性矩阵采用重启随机游走和余弦相似获得药物和靶标的多重相似性特征矩阵,并用PCA实现数据降噪声;然后在异构网络中采用GCN获取药物和靶标的低维表示,运用增强注意力机制获得节点间的注意力,并通过消融实验和对照实验,验证了多重相似性和增强注意力模块的作用.在多个数据集上实验结果表明,本模型达到了较好的性能,提升了DTI预测性能和模型的泛化能力.
参 考 文 献
[1]" 彭利红,田雄飞,周立前.基于一致性学习预测药物-靶标相互作用[J].湖南工业大学学报,2020,34(6):27-33.
PENG L H,TIAN X F,ZHOU L Q.Prediction of drug-target interactions based on consistency learning[J].Journal of Hunan University of Technology,2020,34(6):27-33.
[2]任浩然,邓博韬,李建华,等.药物-靶标相互作用预测平台设计与实现[J].现代计算机,2023,29(5):104-108.
REN H R,DENG B T,LI J H,et al.Design and implementation of drug-target interaction prediction platform[J].Modern Computer,2023,29(5):104-108.
[3]王红梅,郭真俊,张丽杰.基于图神经网络的药物-靶标相互作用预测研究[J].长春工业大学学报,2021,42(4):318-325.
WANG H M,GUO Z J,ZHANG L J.Drug-target interaction prediction based on graph neural network[J].Journal of Changchun University of Technology,2021,42(4):318-325.
[4]YU G X,WANG Y H,WANG J,et al.Attributed heterogeneous network fusion via collaborative matrix tri-factorization[J].Information Fusion,2020,63:153-165.
[5]LUO Y N,ZHAO X B,ZHOU J T,et al.A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information[J].Nature Communications,2017,8:573.
[6]ZHENG X D,DING H,MAMITSUKA H,et al.Collaborative matrix factorization with multiple similarities for predicting drug-target interactions[C]//Proceedings of the 19th ACM SIGKDD international conference on Knowledge discovery and data mining.Chicago:ACM,2013.
[7]WAN F P,HONG L X,XIAO A,et al.NeoDTI:neural integration of neighbor information from a heterogeneous network for discovering new drug-target interactions[J].Bioinformatics,2019,35(1):104-111.
[8]SHANG Y F,YE X C,FUTAMURA Y,et al.Multiview network embedding for drug-target Interactions prediction by consistent and complementary information preserving[J].Briefings in Bioinformatics,2022,23(3):bbac059.
[9]TIAN Z,PENG X Y,FANG H C,et al.MHADTI:predicting drug–target interactions via multiview heterogeneous information network embedding with hierarchical attention mechanisms[J].Briefings in Bioinformatics,2022,23(6):bbac434.
[10]SHAO K H,ZHANG Y H,WEN Y Q,et al.DTI-HETA:prediction of drug-target interactions based on GCN and GAT on heterogeneous graph[J].Briefings in Bioinformatics,2022,23(3):bbac109.
[11]NASCIMENTO A C A,PRUDNCIO R B C,COSTA I G.A multiple kernel learning algorithm for drug-target interaction prediction[J].BMC Bioinformatics,2016,17:46.
[12]ZHANG L L,OUYANG C P,HU F Y,et al.Relational topology-based heterogeneous network embedding for predicting drug-target interactions[J].Data Intelligence,2023,5(2):475-493.
[13]LU Z L,WANG Y K,ZENG M,et al.HNEDTI:Prediction of drug-target interaction based on heterogeneous network embedding[C]//2019 IEEE International Conference on Bioinformatics and Biomedicine(BIBM).[S.l.]:IEEE,2019:211-214.
[14]廖懿鸣,欧阳纯萍,刘永彬,等.基于异质信息网络元路径的药物-靶标相互作用预测模型[J].北京大学学报(自然科学版),2022,58(1):37-44.
LIAO Y M,OUYANG C P,LIU Y B,et al.Drug-target interactions prediction based on meta-path of heterogeneous information network[J].Acta Scientiarum Naturalium Universitatis Pekinensis,2022,58(1):37-44.
[15]彭利红,刘海燕,任日丽,等.基于多标记学习预测药物-靶标相互作用[J].计算机工程与应用,2017,53(15):260-265.
PENG L H,LIU H Y,REN R L,et al.Predicting drug-target interactions with multi-label learning[J].Computer Engineering and Applications,2017,53(15):260-265.
[16]HATTORI M,OKUNO Y,GOTO S,et al.Development of a chemical structure comparison method for integrated analysis of chemical and genomic information in the metabolic pathways[J].Journal of the American Chemical Society,2003,125(39):11853-11865.
[17]SMITH T F,WATERMAN M S.Identification of common molecular subsequences[J].Journal of Molecular Biology,1981,147(1):195-197.
[18]MA Z H,KUANG Z F,DENG L.CRPGCN:predicting circRNA-disease associations using graph convolutional network based on heterogeneous network[J].BMC Bioinformatics,2021,22(1):551.
[19]HUANG D,AN J Y,ZHANG L,et al.Computational method using heterogeneous graph convolutional network model combined with reinforcement layer for MiRNA-disease association prediction[J].BMC Bioinformatics,2022,23(1):299.
[20]LAW V,KNOX C,DJOUMBOU Y,et al.DrugBank 4.0:shedding new light on drug metabolism[J].Nucleic Acids Research,2014,42(Database issue):D1091-D1097.
[21]SCHOMBURG I,CHANG A,PLACZEK S,et al.BRENDA in 2013:integrated reactions,kinetic data,enzyme function data,improved disease classification:new options and contents in BRENDA[J].Nucleic Acids Research,2013,41(Database issue):D764-D772.
[22]KANEHISA M,GOTO S,HATTORI M,et al.From genomics to chemical genomics:new developments in KEGG[J].Nucleic Acids Research,2006,34(Database issue):D354-D357.
[23]HECKER N,AHMED J,VON EICHBORN J,et al.SuperTarget goes quantitative:update on drug-target interactions[J].Nucleic Acids Research,2012,40(Database issue):D1113-D1117.
[24]LI Y,QIAO G Y,WANG K Q,et al.Drug-target interaction predication via multi-channel graph neural networks[J].Briefings in Bioinformatics,2022,23(1):bbab346.
[25]PENG J J,LI J Y,SHANG X Q.A learning-based method for drug-target interaction prediction based on feature representation learning and deep neural network[J].BMC Bioinformatics,2020,21(Suppl 13):394.
[26]LIU Y,WU M,MIAO C Y,et al.Neighborhood regularized logistic matrix factorization for drug-target interaction prediction[J].PLoS Computational Biology,2016,12(2):e1004760.
[27]PLIAKOS K,VENS C.Drug-target interaction prediction with tree-ensemble learning and output space reconstruction[J].BMC bioinformatics,2020,21(2):1-11.
Prediction of drug-target interaction based on multiple similarity and enhanced attention mechanisms on graph convolution neural network
Wang Wei1a,b, Yu Mengxue1a, Sun Bin1a, Wan Shitong1a, Liu Dong1a,b, Zhou Yun1a,b, Zhang Hongjung2, Wang Xianfang3
(1. a. College of Computer and Information Engineering;" b. Key Laboratory of Artificial Intelligence and Personalized Learning in Education
of Henan Province, Henan Normal University, Xinxiang 453007, China; 2. Hebi Instiute of Engineering and Technology,
Henan Polytechnic University, Hebi 458030, China; 3. College of Computer Science and Technology Engineering,
Henan Institute of Technology, Xinxiang 453000, China)
Abstract: In the research of new drug discovery and drug repositioning, it is important to search for the interactions between drugs and targets. In this study, we propose a graph convolutional network model based on multiple similarities and enhanced attention mechanism to predict drug-target interactions(RSGCN) for the drug-target interaction network. Firstly, we propose to use multiple similarities to optimize the original feature vectors of drugs and targets, capture network structure features, and fully utilize direct or indirect relationships between nodes. Then, we reduce the impact of similarity noise on experimental results through PCA dimensionality reduction. Finally, we use GCN to obtain node embedding representations and incorporate an attention-based enhanced layer to obtain attention weights between nodes, which efficiently predicts interactions between drugs and targets. The experiments use a public gold standard dataset, and the experimental results indicate that the RSGCN model has good performance.
Keywords: graph convolution neural network; multiple similarity; PCA; enhanced attention mechanisms; drug-target interaction
[责任编校 陈留院 杨浦]
猜你喜欢
中老年保健(2022年3期)2022-08-24 02:58:40
中老年保健(2022年1期)2022-08-17 06:14:54
中老年保健(2021年9期)2021-08-24 03:50:50
中老年保健(2021年12期)2021-08-24 03:31:20
新疆农垦科技(2014年9期)2014-02-28 19:21:04
祝您健康(1984年6期)1984-12-30 06:51:10
