
丹江流域干支流浮游细菌群落结构特征与驱动因子
2025-03-04 00:00:00李娟薛旭东杜豆常朝
河南师范大学学报(自然科学版) 2025年2期





摘 要:浮游细菌是河流生态系统中元素循环和能量流动的主要贡献者,其群落组成和多样性因其地理位置而异.干流和支流通常在水文、地形和人类活动强度方面存在差异,目前对干流和支流之间浮游细菌群落组成的差异比较及其构建机制尚不明确.通过采集丹江流域干流和支流19个样点的浮游细菌样本,进行16S rRNA基因测序和水质分析,确定了丹江流域干流和支流浮游细菌的基本分布格局及其关键环境因子.结果表明,丹江流域浮游细菌主要以变形菌门、放线菌门、拟杆菌门和蓝细菌门为主.丹江支流浮游细菌的α多样性高于干流,并且干流和支流浮游细菌在β多样性组成上存在显著差异.干流浮游细菌群落组成主要受到TOC(总有机碳)和DO(溶解氧)的影响,而支流浮游细菌则主要受TN(总氮)、NO-3-N(硝态氮)和T(水温)的影响.此外,中性群落模型表明丹江干流浮游细菌主要受到确定性过程的影响,而支流主要受到随机性过程的影响.
关键词:丹江流域;浮游细菌;群落构建;生物地理学;驱动因子
中图分类号:Q89""""" 文献标志码:A文章编号:1000-2367(2025)02-0014-10
浮游生物作为水域生态系统中的关键类群,在水域生态系统的生物地球化学循环和水生态健康维持方面发挥着重要作用[1-3].其中,浮游细菌是数量最多、种类最丰富的类群[4-5].浮游细菌群落结构和功能在一定程度上决定着水生态系统的健康和稳定[6-7].因此,明确浮游细菌的分布格局、主要影响因子和群落构建机制,对于理解和保护水域生态系统至关重要[8-9].
丹江作为南水北调中线工程的重要水源,其水质健康对于保障供水安全十分重要.研究丹江的浮游细菌的组成和分布,有助于优化水资源管理,确保南水北调工程的顺利实施和长期稳定运行[10].由于丹江支流河段水量小、地势陡峭、人类活动少,其上游流域以森林和草地等土地利用为主,浮游细菌多来自上游附着于底质的细菌和森林、草地等土壤的输入[11].相反,干流河段水量大、地势平缓、人类活动多,周边土地通常以农田和城市用地为主,浮游细菌则主要来自各支流输入、污水排放等[10,12].因此,丹江干流和支流在水文、地形、人类活动强度等方面的差异能够在一定程度上塑造出不同类型的浮游细菌[6,8-9].目前对于丹江干流和支流浮游细菌群落的组成和多样性之间具体的差异仍然认识不足.水温、pH、氮、磷和盐度是影响浮游细菌群落结构的关键因子[13].然而,丹江干流和支流的浮游细菌是否受相同环境因素驱动尚不明确.此外,尽管环境因子解释了浮游细菌群落组成的大部分变异,但仍有相当一部分未能被解释.近年来,群落构建理论的提出为解决这一问题提供了新的方法[14].确定性和随机性过程是塑造浮游细菌群落的关键机制.确定性过程涉及非随机的生态位机制,如环境过滤和种间相互作用[15].而随机过程则涉及物种相对丰度的随机变动,如随机的
收稿日期:2024-03-21;修回日期:2024-07-20.
基金项目:国家自然科学基金(51979236).
作者简介(通信作者):李娟(1983-),女,陕西神木人,陕西省环境调查评估中心高级工程师,研究方向为环境科学及水生态环境调查评估,E-mail:sxshjdcpgzxlj@163.com.
引用本文:李娟,薛旭东,杜豆,等.丹江流域干支流浮游细菌群落结构特征与驱动因子[J].河南师范大学学报(自然科学版),2025,53(2):14-23.(Li Juan,Xue Xudong,Du Dou,et al.Characterization and driving factors of bacterioplankton community structure in the mainstream and tributaries of the Danjiang River Basin[J].Journal of Henan Normal University(Natural Science Edition),2025,53(2):14-23.DOI:10.16366/j.cnki.1000-2367.2024.03.21.0001.)
出生-死亡事件、概率性的扩散事件以及不可预测的扰动[16].干流和支流在水文、地形和营养盐方面的差异导致了确定性和随机性过程对浮游细菌贡献的比例不同.因此,丹江干流和支流浮游细菌群落的构建机制仍需进一步研究.
了解丹江水体浮游细菌的分布格局、主要影响因子以及群落构建机制,对区域水资源保护具有重要意义.本研究通过采集丹江干流及其7条支流的浮游细菌样本并测定水体理化性质,旨在回答以下问题:1)丹江干流和支流浮游细菌群落组成和多样性的分布格局是什么?2)影响丹江浮游细菌的主要水质因子有哪些?影响干支流浮游细菌的水质因子是否一致?3)丹江浮游细菌群落构建过程是由确定性还是随机性过程占主导?本研究拟通过研究丹江流域干流和支流浮游细菌群落多样性、组成、关键水质因子以及群落构建过程,为明确丹江流域河流浮游细菌的地理分布格局提供新的认识.
1 材料方法
1.1 研究区域概述
丹江作为汉江最大的支流,源自秦岭南麓,流经陕西、河南和湖北3省,最终汇入丹江口水库.其流域范围位于109°30′~111°30′E,32°30′~34°30′N,海拔在100~2 164 m之间,流域总面积达1.07万 km2.流域内涵盖多条支流,包括滔河、银花河和武关河等,总长度约为430 km.研究区的气候类型属于亚热带与温带混合型气候,多年平均气温在11~14 ℃之间.年平均降雨量为791 mm,主要集中在7至9月,高山区降水最多,而河谷川道地区降水相对较少.
1.2 样品采集与分析
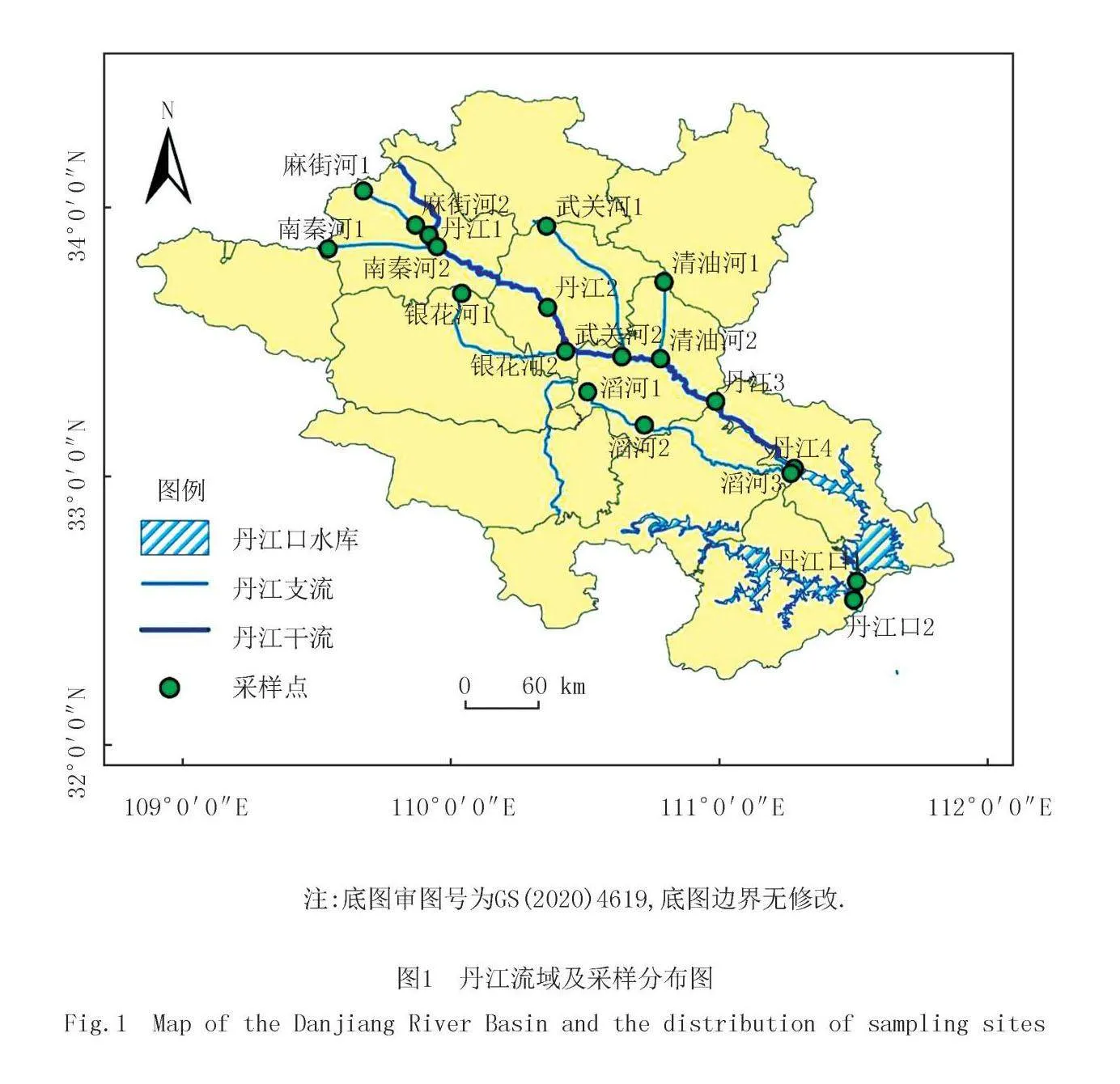
本研究于2023年3月开展丹江流域实地调查与样品采集.共选取丹江干流6个点位,支流14个点位,支流样点包括麻街河3个点位、南秦河2个点位、银花河2个点位,武关河2个点位,清油河2个点位以及滔河3个点位(图1).
使用多参数水质分析仪(Hydrolab HL7,HACH,USA)现场测定pH、DO(溶解氧)、T(水温)、Conductivity(电导率)、Salinity(盐度)、TDS(总溶解性固体)、ORP(氧化还原电位)等水质指标.Turbidity(浊度)使用浊度仪进行测定.使用2 L规格的采水器采集水面下30 cm处水样,样品采集后盛装于500 mL的无菌聚氯乙烯瓶中.所有监测断面均采集3个平行.样品采集完成后放置于车载冰箱内低温避光保存,并迅速运回实验室进行测定.TN(总氮)、TP(总磷)、NH-3-N(氨氮)和NO-3-N(硝氮)按照《水和废水监测分析方法》[17]进行测定,TC(总碳)、IC(无机碳)和TOC(总有机碳)使用总有机碳分析仪(TOC-L CPN,Shimadzu,Japan)进行测定.
在采样后的12 h内,每个点位的1 L水样通过便携隔膜真空泵(无油)过滤,滤膜使用0.22 μm孔径的聚碳酸酯膜过滤器(直径47 mm,Millipore),待回到实验室后存放在-80 ℃,直到进一步的分析.
本研究使用DNeasy PowerSoil试剂盒来提取水体中的DNA.使用通用引物515F(5'-GTGYCAGCMGCCGCGGTAA-3')[18]和926R(5'-CCGYCAATTYMTTTR AGTTT-3')[19]来扩增原核16S rRNA基因.采用通用引物组515F-926R对16S rRNA基因V4-V5区域进行PCR扩增,该引物对在微生物扩增子高通量测序中得到广泛应用[20-21].PCR反应混合物总体积为30 μL,包括15 μL NEB Next Ultra II Q5 Mix(NEB,美国)、3 μL前引物(10 μmol/L)、3 μL后引物(10 μmol/L)、1 μL模板DNA和8 μL无核酸水.PCR反应程序包括98 ℃的初始变性30 s,然后进行32个循环,其中98 ℃变性10 s,56 ℃退火20 s,72 ℃延伸30 s,最终延伸72 ℃ 2 min.PCR产物通过使用Axyprep DNA凝胶提取试剂盒进行纯化.最后,由上海凌恩生物科技有限公司使用Illumina NovaSeq 6000测序.
1.3 生物信息学分析
本研究中的序列数据使用QIIME 2(版本2022.2)进行分析.首先,使用q2-demux插件对原始序列数据进行质控.随后使用DADA2进行去噪并生成扩增子序列变体(ASVs),然后通过q2-dada2生成代表性序列[22].使用q2-feature-classifier中的classify-sklearn朴素贝叶斯分类器,使用默认参数,根据SILVA v138 16S rRNA基因数据库参考序列对ASV进行分类[23].注释完成后,手动剔除未注释的ASVs序列.
1.4 统计分析
使用vegan包计算浮游细菌群落α多样性,干流和支流的差异分析使用独立样本t检验,判断阈值为p<0.05.使用基于Bray-Curtis距离的非度量多维尺度分析(NMDS)评估浮游细菌组成差异,两个分组(干流和支流)之间的差异使用ANOSIM进行检验.浮游细菌门水平或属水平类群和环境因子之间的相关性分析使用Spearman相关性进行分析.使用envfit函数筛选与浮游细菌显著相关的环境因子,随后进行冗余分析(RDA).此外,使用RandomForest包进行随机森林分析,以识别干流和支流各自重要的水质因子,在进行随机森林分析之前,首先对浮游细菌进行了主成分分析(PCA),通过R语言中vegan包的rda函数进行计算.此外,使用中性群落模型研究了这些群落组装中随机过程的潜在重要性,其中m值估计群落间扩散,R平方值量化随机过程的贡献[24].
2 结果与分析
2.1 浮游细菌的群落组成及多样性
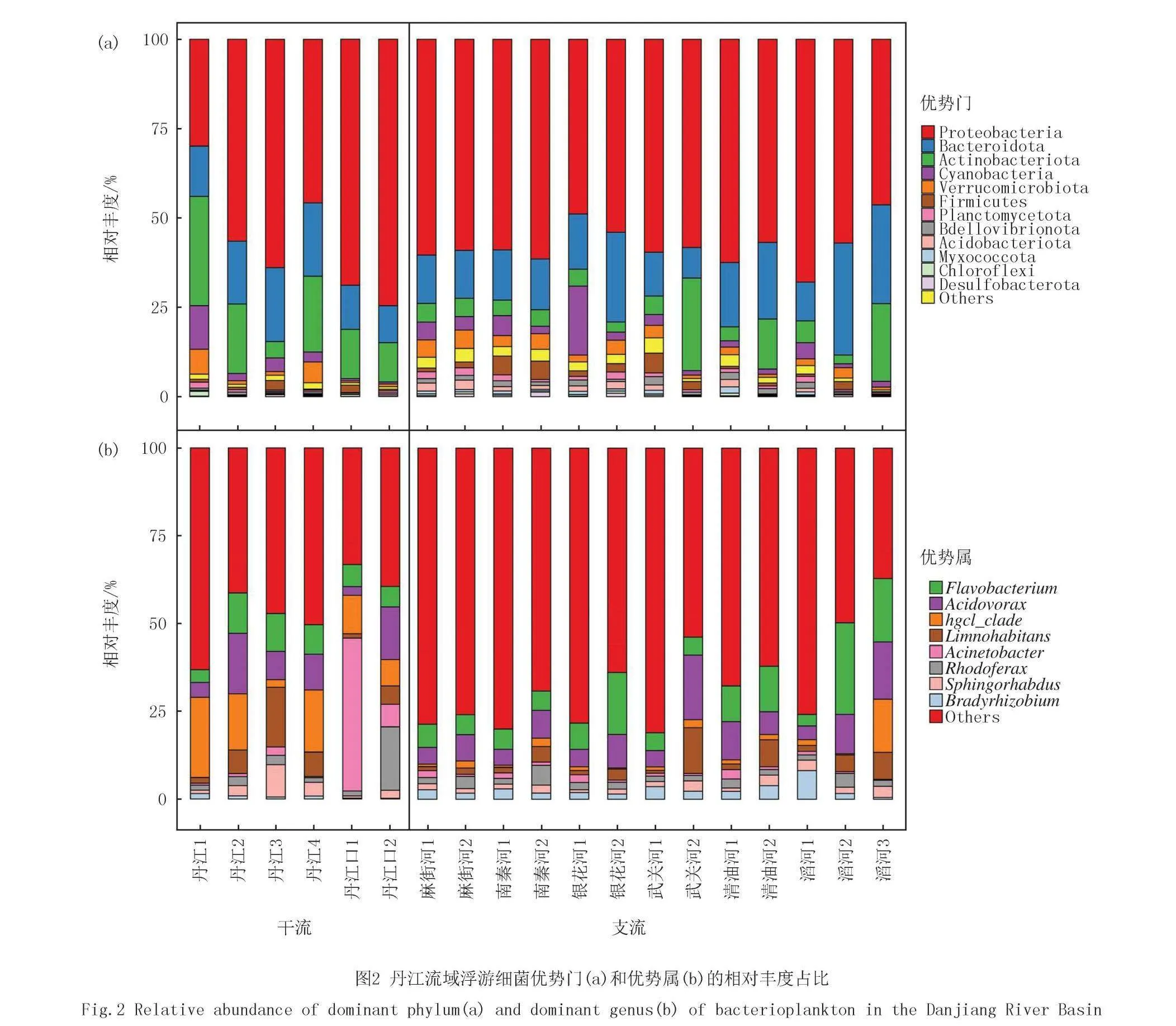
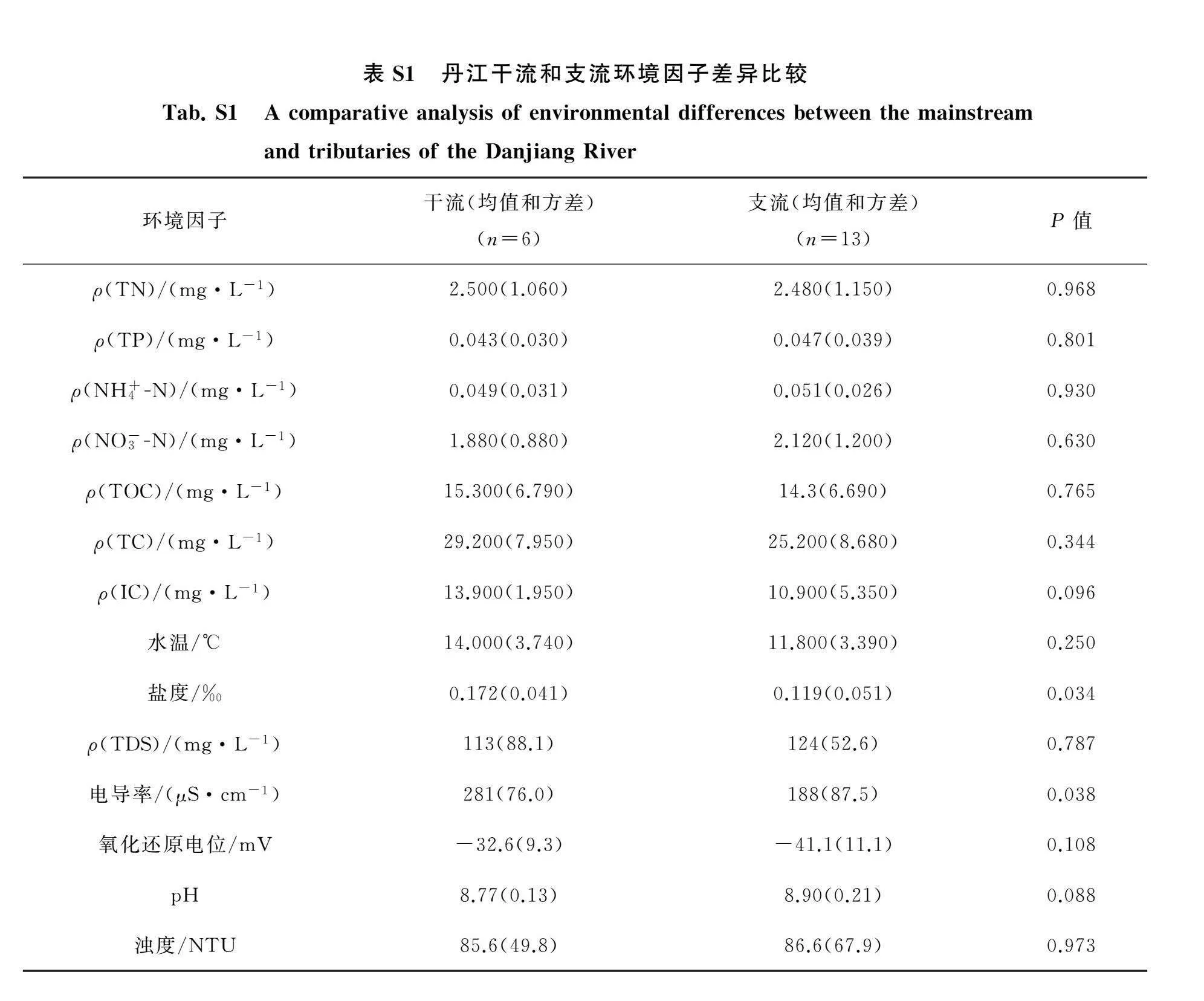
本研究通过比较丹江干流和支流的水质因子发现除盐度和电导率之外,其他环境因子无显著差异(附录表S1).对于浮游细菌而言,在进行后续分析之前,对每个样本的序列进行了抽平处理,抽平数为56 185,最终保留了19 455条ASVs和1 067 515条有效序列数.在门水平上(图2(a)),优势门及其在干流和支流的相对丰度分别为Proteobacteria(变形菌门,干流55.57%,支流57.78%)、Actinobacteriota(放线菌门,干流16.78%,支流8.17%)、Bacteroidota(拟杆菌门,干流15.92%,支流17.37%)、Cyanobacteria(蓝细菌门,干流3.67%,支流4.05%)、Verrucomicrobiota(疣微菌门,干流2.68%,支流2.80%)、Firmicutes(厚壁菌门,干流1.15%,支流2.27%)、Chloroflexi(绿弯菌门,干流0.47%,支流0.37%)、Desulfobacterota(脱硫菌门,干流0.18%,支流0.43%)、Myxococcota(粘球菌门,干流0.47%,支流0.37%)、Acidobacteriota(酸杆菌门,干流0.08%,支流1.25%).
在属水平上(图2(b)),优势属及其在干流和支流的相对丰度分别为hgcI_clade(hgcI类群,干流9.54%,支流8.56%)、Acidovorax(食酸菌属,干流9.54%,支流8.56%)、Acinetobacter(不动杆菌属,干流9.03%,支流1.13%)、Flavobacterium(黄杆菌属,干流7.74%,支流9.93%)、Limnohabitans(淡水栖菌属,干流6.47%,支流3.93%)、Rhodoferax(红育菌属,干流4.89%,支流5.15%)、Sphingorhabdus(鞘脂杆菌属,干流3.31%,支流1.93%)、Bradyrhizobium(慢生根瘤菌属,干流0.71%,支流2.68%)
图3(a)的结果表明,丹江干流和支流不同河段浮游细菌在门水平上的相对丰度差异明显.其中,变形菌门从干流上游到下游有增加的趋势.而不同样点之间属水平的群落组成差异相较于门水平更加明显(图2).特别是丹江干流和支流浮游细菌优势属的占比差异十分明显(图3(b)).丹江干流的hgcl_clade(hgcl类)、Acinetobacter(不动杆菌属)和Sphingorhabdus(鞘脂杆菌属)的相对丰度远高于支流,而Flavobacterium(黄杆菌属)、Rhodoferax(红育菌属)和Bradyrhizobium(慢生根瘤菌)则是支流的相对丰度高于干流.
通过对丹江干流和支流浮游细菌的α多样性进行比较(图4),发现支流的ACE、Pielou evenness、Richness以及Shannon多样性都显著高于干流.此外,通过NMDS发现干流和支流之间的浮游细菌在排序轴上有明显的区分,且通过Anosim分析发现干流和支流基于Bay-Curtis距离的浮游细菌群落之间存在显著差异,反映了干流和支流浮游细菌β多样性存在明显的差异(图5).
2.2 浮游细菌的关键环境驱动因子
总氮和硝氮与除Actinobacteria(放线菌门)和Deinococcota(奇异球菌门)外的其他门类均呈现出明显的正相关关系,而水温则与之呈现出明显的负相关关系(图6(a)).
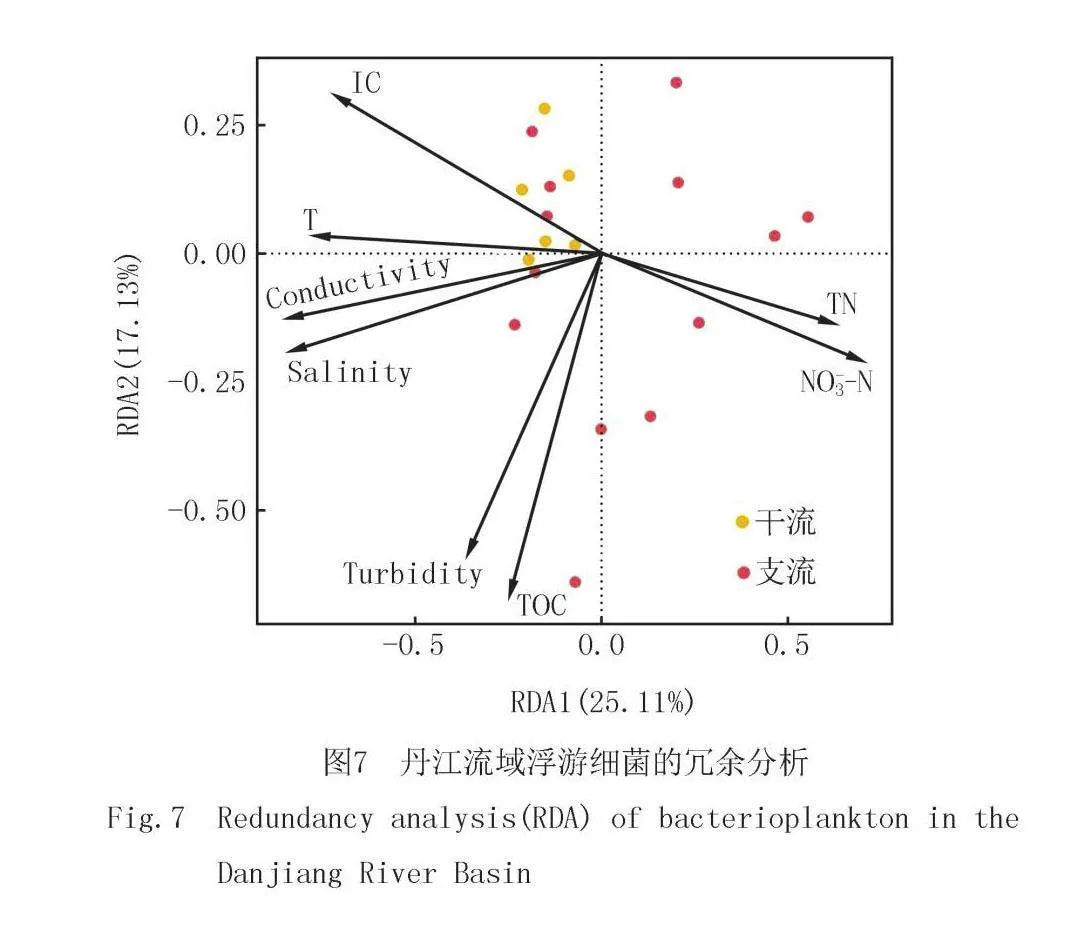
通过冗余分析发现总氮、温度、总有机碳、溶解氧和浊度是影响丹江浮游细菌的主要环境因子,并且通过RDA分析解释率达到了44%(图7).此外,通过随机森林明确了环境因子对干流和支流浮游细菌群落的主成分1和主成分2的解释量(图8),发现干流浮游细菌主要受到了Actinobacteria(放线菌门)和Deinococcota(奇异球菌门)和溶解氧的影响,而对于支流浮游细菌而言其主要受到了总氮、温度以及硝态氮的影响,干流和支流的浮游细菌群落受到的主要水质因子存在明显差异.
2.3 浮游细菌的群落构建过程
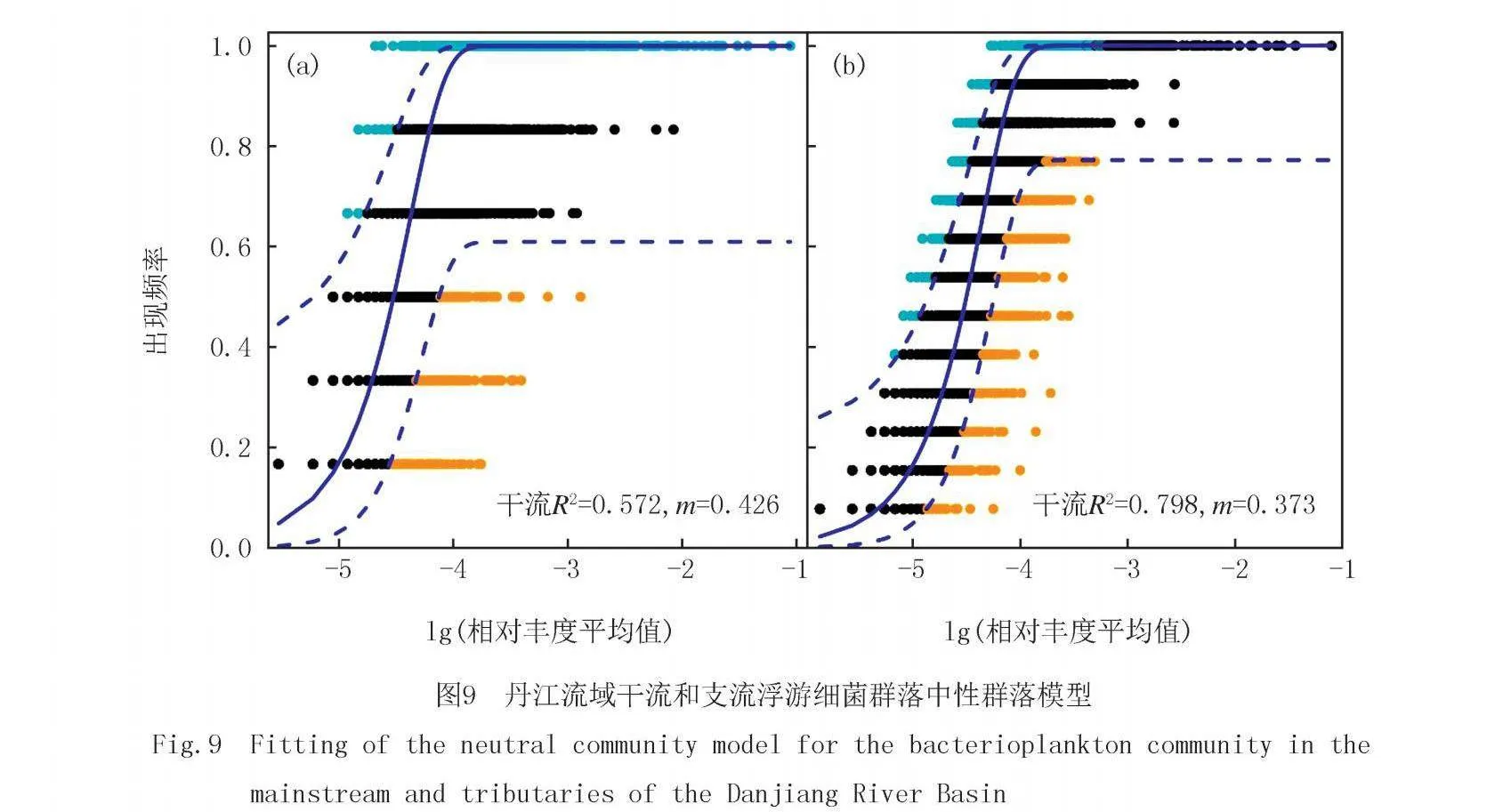
中性群落模型能够用来估计生态随机性.其中,R2>0.5认为群落主要受到随机性过程的影响,而R2<0.5则表明群落主要受到确定性过程的影响.本研究发现,干流的R2为0.572而支流的R2为0.798(图9),干流和支流的群落构建都以随机性过程为主导,但是支流的随机性过程更高.此外,通过m值发现干流的扩散能力要高于支流.
3 讨 论
3.1 丹江干流和支流浮游细菌群落组成和多样性比较
本研究结果表明丹江干流与支流浮游细菌优势属占比差异明显.其中,丹江干流的hgcl_clade(hgcl类群)、Acinetobacter(不动杆菌属)和鞘脂杆菌属(Sphingorhabdus)的相对丰度远高于支流,而Flavobacterium(黄杆菌属)、Rhodoferax(红育菌属)和Bradyrhizobium(慢生根瘤菌属)则是支流的相对丰度高于干流.此外,本研究发现丹江支流浮游细菌的多样性要高于干流.首先,干流和支流各自距河流源头的距离差异直接影响了浮游细菌的生长繁殖.支流的浮游细菌从源头到支流下游的水力停留时间较短,而干流的浮游细菌从源头到干流下游的水力停留时间长,这使得干流中适应淡水环境的细菌能够生长繁殖,而以冲刷方式进入水体的土壤细菌中不耐受水环境的细菌会逐渐减少.有研究对泰晤士河流域上游支流以及下游主干道的水样浮游细菌群落分析发现了河流浮游细菌从上游以拟杆菌门主导转变为下游以放线菌门主导的生态演替[18].由于Flavobacterium(黄杆菌属)和Bradyrhizobium(慢生根瘤菌属)主要生活在土壤中,而Acinetobacter(不动杆菌属)和鞘脂杆菌属(Sphingorhabdus)则广泛存在于水体和土壤环境中,这使得丹江浮游细菌群落从支流以Flavobacterium(黄杆菌属)和Sphingorhabdus(鞘脂杆菌属)主导转变为干流以hgcl_clade(hgcl类群)和Acinetobacter(不动杆菌属)为优势属.
其次,研究区域支流受到的人类活动干扰小于干流.这也是造成干流和支流浮游细菌不同的重要因素,同时也使得干流浮游细菌的多样性低于支流.本研究中支流基本处于海拔较高的山地峡谷内,其周边的土地主要为森林和草地,而干流则处于地势相对平缓的地区,河流经过的区域以农田为主,并经过多个城镇.大量研究表明人类活动强度的增加会导致河流生物多样性的下降[25-27].有研究对亚热带气候区的河流进行调查发现,随着农田和城市用地的增加,下游水域中的水生生物多样性呈现出明显的下降趋势[27].因此,人类活动也是造成干流和支流浮游生物群落组成和多样性具有差异的重要因素.
此外,干流和支流的浮游细菌的来源具有明显的差异.相较于干流而言,支流上游底质附着细菌对支流下游浮游细菌群落的贡献占比更高.此外,支流上游相较于干流具有更强的水陆连通性,这有助于浮游细菌从周边土壤及地下水迁徙[28].而干流中的浮游细菌往往受到不同来源支流汇入的影响.干流不同河段会受纳不同支流的浮游细菌,并且在这一过程中会发生群落融合现象[28].当干流和支流的水环境差异比较大时,会产生比较强的异质性选择作用(异质的环境条件导致群落结构的相似性减少),这会造成干流中浮游细菌的多样性下降[28].此外,干流河流相较于支流会有更多的污水处理厂尾水的输入,这也是造成干流浮游细菌群落改变的重要因素.因此,研究区域干流和支流浮游细菌群落组成和多样性的差异是水文、地形、土地利用类型和人类活动综合作用的结果.
3.2 水质对浮游细菌群落的影响
本研究通过分析优势门与环境因子的相关性发现(图6a)总氮和硝氮与除放线菌门和Deinococcota(奇异球菌门)外的其他门类均呈现出明显的正相关关系,而水温则与之呈现出明显的负相关关系.此外,属水平上也能够发现类似的结果,即总氮与多数属呈正相关,而温度呈负相关.氮是生物体中蛋白质的重要构成元素,总氮浓度在一定范围内的增加能够促进浮游细菌的生长与繁殖.有研究通过室内模拟不同氮水平下微生物群落的变化,发现当水体中的总氮质量浓度从1 mg/L增加到5 mg/L时,微生物的代谢活性增加了约30%,生物量(以总有机碳计)增加了20%[29].此外,在高氮水平(5 mg/L)下,Proteobacteria(变形菌门)和Bacteroidetes(拟杆菌门)的相对丰度分别增加了15%和10%.这表明总氮的增加显著促进了这些优势门类的生长.本研究中发现Actinobacteria(放线菌门)和Deinococcota(奇异球菌门)与总氮呈现负相关.其中,Actinobacteria(放线菌门)在有机质分解中扮演重要角色,其在水体中的丰度可能更多地受到有机质含量的影响,而不是直接依赖于氮素水平.Deinococcota(奇异球菌门)并没有足够的参考资料.
本研究发现水温增加使得除Actinobacteria(放线菌门)和Deinococcota(奇异球菌门)外的其他门类相对丰度降低.有研究对潮白河进行季节性调查发现,当水体温度从20 ℃升高到30 ℃时,微生物的总生物量减少了约25%,同时变形菌门和拟杆菌门的相对丰度分别下降了12%和8%[30].这一结果表明高温对浮游细菌中的一些门类的生长具有抑制作用.该研究还发现高温可能加速了氮循环过程中的反硝化作用,从而降低了水体中的总氮和硝氮浓度.综上所述,本研究研究区域总氮和硝氮的增加促进了多数浮游细菌门类的增加,而较高的温度则降低了多数浮游细菌门类的相对丰度,并使得总氮和硝氮的浓度下降.
进一步的研究表明,干流和支流浮游细菌群落受到的主要环境因子存在显著差异.干流浮游细菌主要受总有机碳和溶解氧的影响,而支流浮游细菌则主要受温度、总氮和硝态氮的影响.本研究中从各支流到干流下游,水体中的总有机碳含量呈现出增加的趋势,而水体中的溶解氧有下降的趋势,这主要是由于下游地区人类活动强度增加导致的.干流水体总有机碳的增加和溶解氧的下降对浮游细菌群落结构的变化产生了直接的影响.针对渭河的调查结果显示当河流中的总有机碳从6.4 mg/L增加到15.5 mg/L时,浮游细菌数量显著增加,同时细菌的呼吸速率也明显提升,造成了水体溶解氧的下降[31].相对而言,支流水体水量小,不同支流的温度的变化相对较大,这可能是导致支流浮游细菌对温度敏感的重要原因.此外,支流与河岸土壤之间较强的水陆连通性可能使得河岸带土壤中的氮素更容易排入支流,从而对浮游细菌群落产生影响[32].干流和支流的环境差异主要由流量、营养物质、溶解氧浓度和温度变化等因素引起,这些差异导致了浮游细菌群落结构的显著差异.
3.3 丹江干流和支流浮游细菌群落构建比较
本研究通过中性群落模型拟合发现干流和支流的群落构建过程存在显著差异,干流相较于支流而言受到的确定性过程更高,而支流则以随机性过程为主导.有对长江水体浮游细菌的研究发现干流由于pH、溶解氧等因子的变化导致环境选择和物种竞争等确定性过程对浮游细菌群落的影响较大[33].而支流由于流量较小、环境波动较大,更容易受到漂变和迁移等随机性过程的影响.相较于干流而言,支流的浮游细菌来自上游底质附着细菌的贡献占比更高.此外,支流上游相较于干流具有更强的水陆连通性[33],这有助于浮游细菌从周边土壤及地下水迁徙,这些过程都强调了随机性过程中的扩散作用对支流浮游细菌的影响.而干流则由于人类活动强度的增加导致水体中营养物质的增加和其他水质因子的变化,从而增加了环境选择的作用.
尽管中性群落模型对浮游细菌生物数据的拟合效果较好,但推断环境和空间变量对浮游细菌生物群落组成的影响仍然存在困难,特别是冗余分析揭示的大量未解释方差.这些未解释的方差可能是由漂移、未测量的环境变量和物种相互作用引起的.一些关键环境变量可能以随机方式变化,短时间的采样通常包含大量噪声,这可能掩盖浮游细菌的生态模式.此外,除了随机过程外,本研究未能确定其他重要机制来解释浮游细菌生物群落的分布,例如质量效应(质量效应假设大规模的物种扩散可以补偿微生物之间由于竞争排斥造成的物种多样性下降,从而减缓群落组成与局部环境变量之间的关联)[34].此外,微生物群落之间的共存关系也可能对群落的稳定性产生重要影响.不同分类和功能群的微生物在面对环境胁迫时会产生不同的响应模式.
总之,通过结合确定性和随机性过程,可以更全面地理解干流和支流浮游细菌群落的构建机制.除了考虑环境因子对浮游细菌的影响外,还需考虑中性过程,从而增加对丹江流域浮游细菌群落关键控制因素的理解.这些发现揭示了不同水环境下微生物群落构建的机制和动态,对理解河流生态系统中微生物的空间分布和生态过程具有重要意义.
4 结 论
本研究通过16S rDNA测序结合水质数据分析,发现丹江浮游细菌优势门和属在干支流之间存在明显差异.并且发现丹江支流浮游细菌的α多样性高于干流,且干流和支流的浮游细菌在β多样性组成上存在显著差异.干流和支流浮游细菌群落所受的主要环境因子存在显著差异.此外,干流中浮游细菌主要受总有机碳和溶解氧的影响,而支流中浮游细菌则主要受总氮、硝态氮和水温的影响.干流和支流的环境差异主要由流量、营养物质、溶解氧浓度和温度变化等因素引起,这些差异导致了浮游细菌群落结构的显著不同.并且,丹江干流的浮游细菌群落主要受到确定性过程的影响,而支流则主要受随机性过程的影响.本研究通过比较丹江干流和支流浮游细菌群落组成和群落构建,证实了丹江干流和支流浮游细菌存在明显差异,并且指出了不同的水系结构在浮游细菌群落构建中的关键作用.未来的研究应进一步关注河网差异对水生生物造成的影响,为保护丹江水生态健康提供基础理论支撑.
附录见电子版(DOI:10.16366/j.cnki.1000-2367.2024.03.21.0001).
参 考 文 献
[1] "GAO Y,ZHANG W L,LI Y.Microbial community coalescence:does it matter in the Three Gorges Reservoir?[J].Water Research,2021,205:117638.
[2]李轶,雷梦婷,杨楠,等.河流微生物生态学的研究进展[J].水资源保护,2022,38(1):190-197.
LI Y,LEI M T,YANG N,et al.Research and prospect on river microbial ecology[J].Water Resources Protection,2022,38(1):190-197.
[3]朱爱萍,原升艳,梁作兵,等.亚热带城市河流浮游细菌群落的季节演替及构建机制[J].环境科学学报,2023,43(2):461-473.
ZHU A P,YUAN S Y,LIANG Z B,et al.Seasonal succession and assembly processes of bacterioplankton communities in a subtropical urban river[J].Acta Scientiae Circumstantiae,2023,43(2):461-473.
[4]王力,王丝可,左剑恶,等.基于浮游细菌生物完整性指数的城市河流健康评价:以深圳河流域为例[J].环境工程学报,2023,17(6):2007-2014.
WANG L,WANG S K,ZUO J E,et al.Urban river health assessment based on biotic integrity bacterioplankton-index of biotic integrity:A case study of Shenzhen River Basin[J].Chinese Journal of Environmental Engineering,2023,17(6):2007-2014.
[5]WANG H L,ZHANG W L,LI Y,et al.Hydrodynamics-driven community coalescence determines ecological assembly processes and shifts bacterial network stability in river bends[J].Science of the Total Environment,2023,858:159772.
[6]吴波波,王鹏,丁明军,等.人类活动强度对锦江浮游细菌群落结构的影响[J].环境科学学报,2022,42(8):459-473.
WU B B,WANG P,DING M J,et al.Effects of anthropogenic intensity on bacterioplankton community structure in Jinjiang River[J].Acta Scientiae Circumstantiae,2022,42(8):459-473.
[7]黄祎,王鹏,吴波波,等.鄱阳湖流域锦江水体污染胁迫下浮游细菌群落结构及交互作用[J].微生物学报,2022,62(12):4564-4576.
HUANG Y,WANG P,WU B B,et al.Community structure and interaction of bacterioplankton under pollution stress in Jinjiang River of Poyang Lake basin[J].Acta Microbiologica Sinica,2022,62(12):4564-4576.
[8]朱信政.南水北调东线工程浮游及沉积物细菌空间分布以及种间关系研究[D].西安:西安理工大学,2022.
ZHU X Z.Spatial distribution and interspecific relationship of bacteria in plankton and sediment of the eastern route of south-to-north water transfer project[D].Xi'an:Xi'an University of Technology,2022.
[9]胡愈炘,张静,黄杰,等.长江流域河流和湖库的浮游细菌群落差异[J].环境科学,2022,43(3):1414-1423.
HU Y X,ZHANG J,HUANG J,et al.Characteristics of bacterioplankton community between river and lake/reservoir in the Yangtze River Basin[J].Environmental Science,2022,43(3):1414-1423.
[10]HAN X,PAN B Z,JIN X W,et al.The assembly mechanisms of algal community across different habitats mediated by sediment in the heavily sediment-laden Yellow River[J].Journal of Hydrology,2024,631:130825.
[11]WEST J R,WHITMAN T.Disturbance by soil mixing decreases microbial richness and supports homogenizing community assembly processes[J].FEMS Microbiology Ecology,2022,98(9):fiac089.
[12]PAN B Z,LIU X Y,CHEN Q W,et al.Hydrological connectivity promotes coalescence of bacterial communities in a floodplain[J].Frontiers in Microbiology,2022,13:971437.
[13]CUSTER G F,BRESCIANI L,DINI-ANDREOTE F.Toward an integrative framework for microbial community coalescence[J].Trends in Microbiology,2024,32(3):241-251.
[14]JIAO S,YANG Y F,XU Y Q,et al.Balance between community assembly processes mediates species coexistence in agricultural soil microbiomes across Eastern China[J].The ISME Journal,2020,14(1):202-216.
[15]郑震,冷东梅,郑莺,等.山仔水库环境因子与藻类群落生长的相互作用机制探究及应对建议[J].灌溉排水学报,2023,42(11):131-139.
ZHENG Z,LENG D M,ZHENG Y,et al.The Interplay between Environmental Factors and Algal Community Growth in Shanzai Reservoir[J].Journal of Irrigation and Drainage,2023,42(11):131-139.
[16]CHASE J M,MYERS J A.Disentangling the importance of ecological niches from stochastic processes across scales[J].Biological Sciences,2011,366(1576):2351-2363.
[17]国家环境保护总局水和废水监测分析方法编委会.水和废水监测分析方法[M].4版.北京:中国环境科学出版社,2002.
[18]CAPORASO J G,LAUBER C L,WALTERS W A,et al.Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample[J].Proceedings of the National Academy of Sciences of the United States of America,2011,108(Suppl 1):4516-4522.
[19]PARADA A E,NEEDHAM D M,FUHRMAN J A.Every base matters:assessing small subunit rRNA primers for marine microbiomes with mock communities,time series and global field samples[J].Environmental Microbiology,2016,18(5):1403-1414.
[20]WANG S,HOU W G,JIANG H C,et al.Microbial diversity accumulates in a downstream direction in the Three Gorges Reservoir[J].Journal of Environmental Sciences,2021,101:156-167.
[21]CHEN J,WANG P F,WANG C,et al.Fungal community demonstrates stronger dispersal limitation and less network connectivity than bacterial community in sediments along a large river[J].Environmental Microbiology,2020,22(3):832-849.
[22]CALLAHAN B J,MCMURDIE P J,ROSEN M J,et al.DADA2:High-resolution sample inference from Illumina amplicon data[J].Nature Methods,2016,13(7):581-583.
[23]QUAST C,PRUESSE E,YILMAZ P,et al.The SILVA ribosomal RNA gene database project:improved data processing and web-based tools[J].Nucleic Acids Research,2013,41(D1):D590-D596.
[24]SLOAN W T,LUNN M,WOODCOCK S,et al.Quantifying the roles of immigration and chance in shaping prokaryote community structure[J].Environmental Microbiology,2006,8(4):732-740.
[25]SONG J X,LIANG D.Community structure of zooplankton and its response to aquatic environmental changes based on eDNA metabarcoding[J].Journal of Hydrology,2023,622:129692.
[26]LI F L,ZHANG Y,ALTERMATT F,et al.Destabilizing effects of environmental stressors on aquatic communities and interaction networks across a major river basin[J].Environmental Science amp; Technology,2023,57(20):7828-7839.
[27]LI F L,QIN S,WANG Z Y,et al.Environmental DNA metabarcoding reveals the impact of different land use on multitrophic biodiversity in riverine systems[J].Science of the Total Environment,2023,855:158958.
[28]CHANG C,HU E,SHI Y F,et al.Linking microbial community coalescence to ecological diversity,community assembly and species coexistence in a typical subhumid river catchment in Northern China[J].The Science of the Total Environment,2024,938:173367.
[29]ZOU X,GAO M J,MOHAMMED A,et al.Responses of various carbon to nitrogen ratios to microbial communities,kinetics,and nitrogen metabolic pathways in aerobic granular sludge reactor[J].Bioresource Technology,2023,367:128225.
[30]LIAO K,BAI Y H,HUO Y,et al.Integrating microbial biomass,composition and function to discern the level of anthropogenic activity in a river ecosystem[J].Environment International,2018,116:147-155.
[31]YAN Z W,YANG N,LIANG Z,et al.Active dissolved organic nitrogen cycling hidden in large river and environmental implications[J].Science of the Total Environment,2021,795:148882.
[32]STADLER M,DEL GIORGIO P A.Terrestrial connectivity,upstream aquatic history and seasonality shape bacterial community assembly within a large boreal aquatic network[J].The ISME Journal,2022,16(4):937-947.
[33]LIU T,ZHANG A N,WANG J W,et al.Integrated biogeography of planktonic and sedimentary bacterial communities in the Yangtze River[J].Microbiome,2018,6(1):16.
[34]CHEN W D,REN K X,ISABWE A,et al.Stochastic processes shape microeukaryotic community assembly in a subtropical river across wet and dry seasons[J].Microbiome,2019,7(1):138.
Characterization and driving factors of bacterioplankton community structure in the mainstream and tributaries of the Danjiang River Basin
Li Juan1, Xue Xudong1, Du Dou1, Chang Chao2
(1. Shaanxi Environmental Investigation and Assessment Center, Xi'an 710061, China; 2. College of Natural Resources
and Environment, Northwest Aamp;F University, Yangling 712100, China)
Abstract: Bacterioplankton are major contributors to elemental cycling and energy flow in riverine ecosystems, and their community composition and diversity vary by geographic location. Variations in hydrology, topography, and human activity intensity between mainstreams and tributaries cause different responses of bacterioplankton communities. However, the differences in bacterioplankton community composition and diversity between mainstems and tributaries, as well as their driving factors, are unclear. In this study, we collected bacterioplankton samples from 19 sites in the mainstem and tributaries of the Danjiang River Basin. We identified the basic distribution patterns of bacterioplankton and their key environmental factors in the Danjiang River Basin by using 16S rRNA gene sequencing and water quality analysis. The results showed that planktonic bacteria in the Danjiang River Basin were mainly dominated by Proteobacteria, Bacteroidota, Actinobacteriota and Cyanobacteria. The α-diversity of bacterioplankton was higher in the tributaries compared with that in the mainstem, with a significant difference observed in β-diversity composition between them. Planktonic bacteria in the mainstem were primarily influenced by TOC(total organic carbon) and DO(dissolved oxygen), while those found in tributaries were mainly affected by TN(total nitrogen), NO-3-N(nitrate nitrogen), and T(water temperature). Additionally, neutral community modeling indicated that planktonic bacteria communities within the mainstem of the Danjiang River were primarily influenced by deterministic processes whereas those within tributaries were mainly influenced by stochastic processes. This study emphasizes the key role of drainage structure in shaping bacterioplankton communities.
Keywords: Danjiang River Basin; bacterioplankton; community assembly; biogeography; driving factors
[责任编校 刘洋 赵晓华]
