
机械力作用下牙周膜干细胞调控骨重塑的研究进展
2024-11-06 00:00:00韩行李颖辉李雯雯徐书奎马文盛
国际口腔医学杂志 2024年3期
[关键词] 机械力; 牙周膜干细胞; 成骨分化; 破骨细胞
[中图分类号] R78 [文献标志码] A [doi] 10.7518/gjkq.2024033
正畸牙移动(orthodontic tooth movement, OTM) 取决于力诱导的牙周组织重塑。临床常见缺乏牙周膜结构的粘连牙及种植体无法移动的现象,说明牙周膜在力学诱导的牙周组织重塑中发挥重要作用。牙周膜干细胞(periodontal ligamentstem cell,PDLSC) 作为牙周膜的重要细胞成分,具有自我更新及多向分化能力,可以促进细胞间信号转导,调控免疫并维持组织稳态,在传导机械刺激和组织重塑中发挥重要作用。有研究[1]指出:共培养环境下PDLSC可通过旁分泌途径促进成骨细胞及破骨细胞活动,提示PDLSC在力诱导的骨重塑中发挥了重要作用。本综述系统归纳了机械力刺激作用下PDLSC内调控骨重塑的相关变化,为研究PDLSC将机械信号转化为生物信号进而调控骨重塑的机制提供参考。
1 机械力诱导PDLSC细胞内骨重塑的相关反应
1.1 PDLSC感知机械力刺激
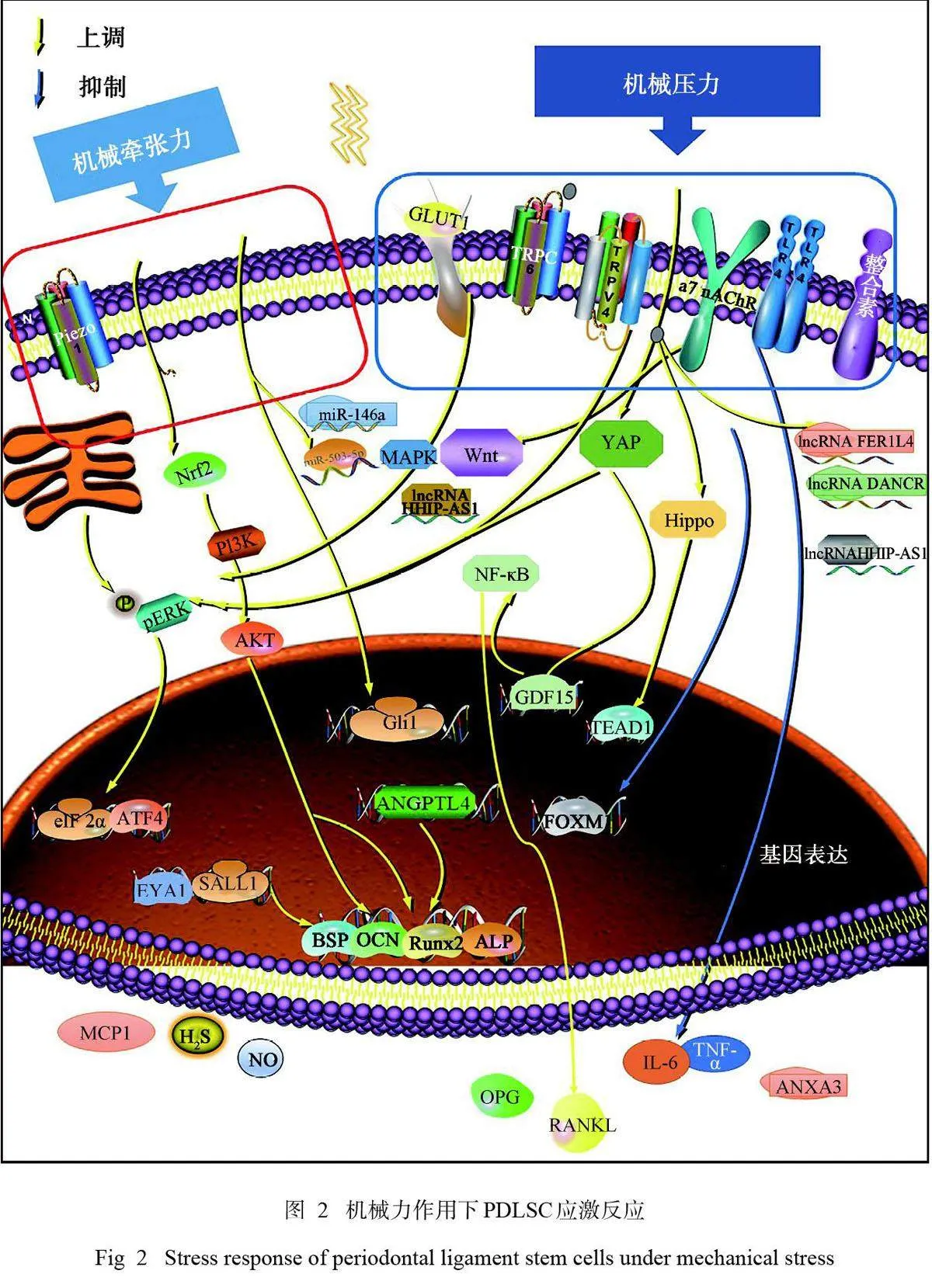
机械力在发育及器官形态发生中至关重要,失重环境会导致宇航员出现显著的骨质流失,说明机械力对骨稳态的调控具有重要意义,骨的形成、吸收、适应都依赖于机械信号[2]。细胞生物力学理论认为:生理状态下人体细胞可受重力、压应力、牵张应力及血流动力学应力等的刺激。对于PDLSC机械力刺激,按照施力类型通常分为压应力和牵张应力,按照施力形式分为静态力和动态循环力。不同形式的作用力对PDLSC的影响不同,而力值大小对细胞反应也有不同程度的影响。机械力学刺激可改变PDLSC的增殖、分化、黏附、迁移、趋化、凋亡、氧化应激;还可诱导PDLSC释放多种骨塑建相关的细胞因子。经典理论[3]认为:PDLSC作为机械敏感细胞在机械力作用下发生一系列的应激反应,细胞信号转导分为4个不同阶段:机械偶联阶段、生化偶联阶段、细胞内信号转导阶段和效应细胞产生反应阶段。施加机械力刺激后,PDLSC通过细胞膜表面机械敏感的通道和受体蛋白完成离子运输和信号转导,并通过第二信使或细胞骨架将机械信号传递到细胞核,或偶联胞内信号通路, 改变细胞状态。此外PDLSC内机械敏感基因的表达、转录因子的调控和非编码RNA的表观遗传学修饰均可受机械力刺激的影响发生变化。
1.2"机械力调控PDLSC成骨向分化
1.2.1"机械力激活细胞膜上的离子通道, 调控PDLSC成骨向分化 1) 瞬时受体电位阳离子通道6 (transient receptor potential channel 6,TRPC6)。适当的机械牵张力激活PDLSC 中的TRPC6,可以加速PDLSC成骨向分化及其对破骨细胞的调控,调控牙周组织重塑,而过度的力学刺激会导致牙槽骨及牙根吸收[4]。2) 压电型机械敏感离子通道组件1 (piezoelectric channel 1,Piezo1) 通道蛋白。牵张应力可促进Piezo1通道蛋白的表达,通过第二信使Ca2+的作用激活Notch1信号通路,促进碱性磷酸酶(alkaline phosphatase,ALP)、Runt相关基因2 (runt-related transcriptionfactor 2,RUNX2)、骨钙素(osteocalcin,OCN)和骨涎蛋白(bone sialoprotein,BSP) 的表达,从而增强其成骨能力[5]。
1.2.2 机械力通过调节细胞应激调控PDLSC 成骨向分化 1) 内质网应激。内质网应激是真核细胞的保护性反应,牵张力可诱导PDLSC中内质网的应激反应,介导激活pERK-eIF2α-ATF4信号通路,从而促进PDLSC的成骨向分化[6]。2) 氧化应激。核红细胞2相关因子2 (nuclear factor erythroid-2 relatedfactor 2,Nrf2) 是抗氧化基因的重要转录激活剂,发挥着重要的抗氧化保护效应[7],它的激活促进周期性牵张应力诱导的PDLSC成骨向分化[8]。
1.2.3 机械力调控发育、分化等相关基因表达调控PDLSC 成骨向分化 1) 干细胞相关基因EYA1(eya transcriptional coactivator and phosphatase 1)及SALL1 (spalt like transcription factor 1)。EYA1是器官特异性干细胞的保守关键调节因子,SALL1 也被认为是干细胞标志物[9]。EYA1 和SALL1是潜在的重要牵张应力敏感基因,以频率依赖性方式参与循环拉伸诱导的PDLSC成骨分化[10]。2) 低密度脂蛋白受体相关蛋白(low-densitylipoprotein receptor,LRP) 6。LRP6是胚胎心脏发育所必需的蛋白, 是经典Wnt 通路的LRP5/LRP6/Frizzled共受体的关键组分,在机械力刺激后的PDLSC中被激活。LRP6缺陷抑制了PDLSC中力依赖性成骨分化和增殖[11]。3) 细胞通讯网络因子1(cellular communication network factor 1,CCN1)。CCN家族在组织纤维化损伤后的修复过程中发挥重要作用,CCN1作为抑制因子可抑制组织纤维化的发生。研究[12] 表明: 机械牵张力刺激诱导PDLSC 向细胞外环境释放CCN1, 从而增强RUNX2和ALP的表达,并促进矿化结节的形成。
1.2.4 机械力调控血管生成相关基因表达介导PDLSC成骨向分化 1) Gli1 (glioma-associated" oncogenehomolog 1) 基因与血管空间分布密切相关,在成血管调控及牵张应力调控PDLSC成骨调控中具有重要作用[13]。2) 血管生成素样因子(angiopoietinlikes,ANGPTL) 是具有调节血管生成作用的分泌性糖蛋白家族。ANGPTL2、3、4主要通过调整糖脂代谢并影响血管生成等影响动脉硬化产生,压应力可诱导ANGPTL4的表达升高,且参与调控PDLSC的成骨向分化[14],但具体机制尚不清楚。
1.2.5 机械力通过非编码RNA调控PDLSC成骨向分化 1) 与肿瘤增殖、侵袭、转移相关的非编码RNA。①miR-146a与自身免疫性疾病和肿瘤的侵袭和转移相关,循环牵张应力下,PDLSC中miR-146a和miR-34a下调,从而增强ALP活性和成骨标志物的表达;同时miR-34a、miR-146a通过调控CUG三联体重复RNA结合蛋白1 (CUG triplet repeatRNA binding protein 1) 类家族成员3 (CUGBPelav-like family member 3,CELF3) 的表达,抑制周期性牵张力诱导的PDLSC成骨分化[15]。②长链非编码RNA (long non-coding RNA, lncRNA)HHIP-AS1在非小细胞肺癌增殖、迁移和侵袭中发挥重要作用。压应力作用下,PDLSC中HHIP-AS1下调,促进成骨分化潜能,抑制PDLSC的迁移和趋化能力[16]。③miR-503-5p与肿瘤增殖密切相关,在大鼠牙移动过程中对牵张力侧牙槽骨形成起负性调控作用[17]。2) 其他微小RNA。miR-3198受压应力刺激上调,受牵张应力作用下调,且miR-3198下调牙周膜细胞中骨保护素(osteoprotegerin,OPG) 的表达[18],抑制了PDLSC的成骨向分化能力。miR-21在机械牵张应力诱导的PDLSC成骨分化中起促进作用,但具体作用仍值得进一步探索[19]。
1.2.6 机械力诱导PDLSC 气体递质释放调控PDLSC成骨向分化 牵张力刺激下,PDLSC 中一氧化氮和一氧化氮合酶(inducible nitric oxide synthase,iNOS) 的表达水平升高,可促进PDLSC的成骨分化,该过程可能由PI3K/Akt/β-catenin信号通路介导[20]。
1.2.7 机械力诱导PDLSC 自噬调控自身成骨向分化 研究[21]发现:机械牵张力刺激下自噬水平的升高能有效促进PDLSC的成骨向分化,说明自噬在正畸力诱导的PDLSC成骨分化中具有重要作用,为正畸治疗促进牙周组织重塑、加速牙齿移动提供了研究基础。
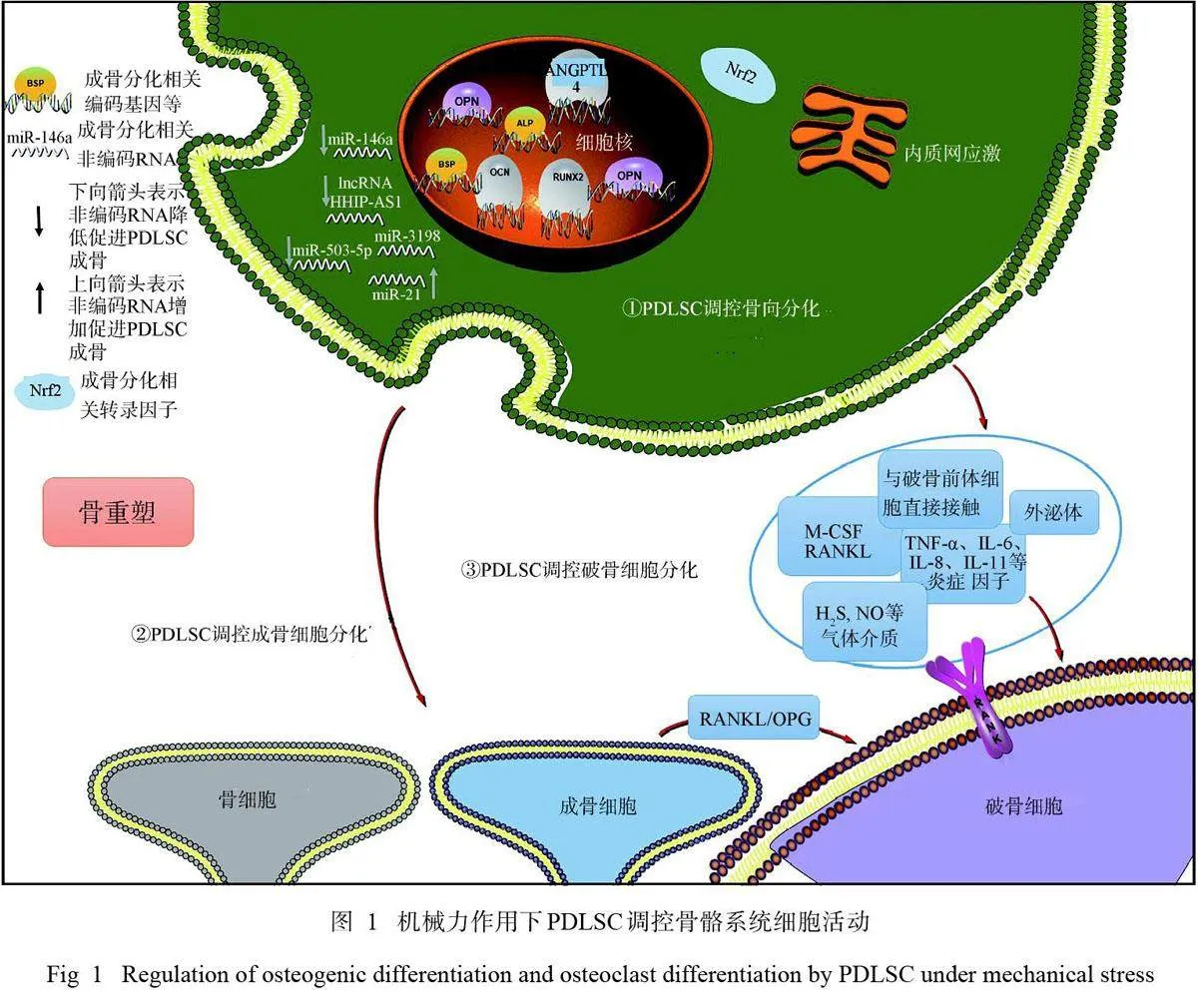
2 机械力作用下PDLSC 通过调控骨骼系统细胞活动影响骨重塑
成骨细胞、破骨细胞、骨细胞作为骨骼系统的主要组成部分,在骨重塑中占据中心环节,其自身可对外界力学刺激发生反应,还受到PDLSC在机械力下的调控作用。此外PDLSC具有成骨向分化潜能,也在骨重塑的骨形成过程中占重要地位。
2.1 机械力作用下PDLSC通过调控成骨细胞活动影响骨重塑
成骨细胞在骨发育、生长和维持中起重要作用[22]。PDLSC-成骨细胞信号通常包括骨形成的刺激因子和抑制因子的表达,许多骨改建调节因子可通过影响成骨细胞的核因子-κB受体激活蛋白配体(receptor activator of nuclear factor- κB ligand,RANKL) 和OPG表达调节破骨细胞的生成[23]。另有研究[1]发现:PDLSC可促进共培养的成骨前细胞MC3T3-E1的ALP活性,增强ALP、BSP、骨桥蛋白(osteopontin,OPN)mRNA和蛋白的表达以及矿化基质的沉积,但具体机制尚不清楚。此外有研究[24] 发现: PDLSC来源的细胞外囊泡(extravesicles,EVs) 可有效增强牵张力作用下成骨细胞内成骨相关蛋白的表达。
2.2 机械力作用下PDLSC与骨细胞相互作用影响骨重塑
骨细胞作为骨骼系统的主要组成部分,是最丰富的成骨细胞谱系细胞,约占成熟骨组织细胞的95%,在骨重塑中起重要作用[25]。机械力作用下,骨细胞与PDLSC之间存在相互作用关系。机械牵张力作用下,骨细胞来源的外泌体通过miR-181b-5p/PTEN/AKT信号通路促进PDLSC增殖,通过BMP2/RUNX2通路促进PDLSC成骨分化,是一种潜在的牙周稳态维持机制[26]。体内实验[27]证明:咬合力刺激通过激活Wnt信号通路上调PDLSC中Gli1的表达,而骨细胞的产物会通过骨硬化蛋白的表达负向调节这一过程。目前有关PDLSC产物对骨细胞作用的研究尚有欠缺,仍需进一步探究。
2.3 机械力作用下PDLSC调控破骨细胞分化
2.3.1 机械力激活PDLSC细胞膜上的机械敏感通道调控破骨细胞分化 1) 瞬时受体电位离子通道4 (transient receptor potential vanilloid 4,TRPV4)机械敏感通道。瞬时受体电位(transient receptorpotential,TRP) 通道调节口腔内稳态,在外部刺激向细胞内信号转导中发挥重要作用[28]。TRPV4是一种与机械刺激和骨代谢相关的钙通道。在体外静压力下,PDLSC表面的TRPV4通道可被激活,并与细胞外信号调控蛋白激酶(extracellular signal-regulated kinase,ERK) 信号通路偶联,导致RANKL及白细胞介素(interleukin,IL) -6、IL-8等炎症相关因子释放增加,促进破骨细胞分化[29]。2) α7烟碱型乙酰胆碱受体(α7 nicotinic acetylcholinereceptor,α7 nAChR) 机械通道。烟碱型乙酰胆碱受体是配体门控的离子通道蛋白,介导突触间信号转导。a7 nAChR是一种特殊异构体,可调节破骨细胞的生成[30]。它可以被尼古丁特异性激活,上调牙周膜细胞中炎症因子的释放,在吸烟的牙周炎患者中研究较广[31]。机械循环压缩应力可以通过上调PDLSC上的α7 nAChR激活典型Wnt信号通路来诱导破骨细胞效应[32]。
2.3.2 机械力激活PDLSC 细胞膜表面受体蛋白调控破骨细胞分化 1) 整合素和钙黏蛋白。整合素是介导细胞与外部环境连接的跨膜受体[33],可与细胞外基质中的其他细胞或配体结合,同时附着于细胞内的细胞骨架上,传递机械力,调节细胞内的信号通路[34]。整合素复合体在PDLSC的压力机械信号接收和转导中发挥作用[35]。avβ3整合素受体在OTM过程中被激活,并与骨吸收功能相关[36]。整合素-黏着斑激酶通路可以调控压力诱导的巨噬细胞集落刺激因子(macrophage colonystimulatingfactor,M-CSF)、肿瘤坏死因子-α (tumornecrosis factor-α,TNF-α)、RANKL和OPG的表达[35]。2) Toll样受体(Toll like receptor,TLR)。TLR具有Ⅰ型整合膜模式识别受体的特征,TLR配体激活间充质干细胞通常会导致促炎和抗炎反应,二者之间的平衡取决于TLR配体的类型和浓度[37]。OTM早期TLR4和IL-1β水平升高,说明TLR参与了早期炎症反应[37]。也有研究[38] 发现激活TLR4可抑制机械压力诱导的IL-6和IL-8的产生。因此,该受体在牙周膜细胞机械力诱导的炎症反应中的双重作用值得进一步探讨。3) 葡萄糖转运蛋白1 (glucose transporter type 1, GLUT1)。GLUT1是一种存在于各种细胞表面的葡萄糖转运蛋白,在糖尿病牙周炎发展机制中具有重要作用。OTM中GLUT1转录被激活,体外实验[39]中压应力激活了PDLSC中的GLUT1,GLUT1敲低抑制了RAW264.7的破骨分化。在大鼠牙齿移动模型研究[40]中,压力侧检测到GLUT1的表达,且GLUT1抑制剂降低了破骨细胞的活性,同时显著降低了小鼠模型的OTM速率。进一步研究[40] 指出:GLUT1的作用与促分裂原活化的蛋白激酶(mitogen-activated protein kinase,MAPK) / ERK信号通路相关,并且对c-Fos信号通路的激活也有影响。上述研究表明:GLUT1的转录激活在机械压应力刺激与骨重塑之间发挥重要的桥梁作用。
2.3.3 机械力通过调节PDLSC 细胞内质网的应激和代谢从而调控破骨细胞分化 1) 生长分化因子(growth differentiation factor, GDF) 15。GDF15是转化生长因子-β (transforming growth factor-β,TGF-β) 超家族成员,与细胞机械反应和破骨细胞活性调控有关[41]。压应力可以通过Yes相关蛋白(yes-associated protein,YAP) 信号分子促进牙周膜细胞中GDF15的表达[42],激活核因子-κB (nuclearfactor-κB,NF-κB) 信号通路,促进ERK磷酸化及炎症因子的释放,增加RANKL/OPG比例,促进RAW264.7破骨分化[43]。此外牵张力也可上调成骨细胞中GDF15的表达,从而促进成骨相关基因的表达。由此可见,GDF15对OTM中的成骨和破骨有双重作用。GDF15作为压力诱导破骨细胞分化的重要介质,在OTM期间PDLSC介导的破骨细胞形成中发挥重要作用,同时对破骨细胞形成的双重调节作用也使其能够调节过度的炎症反应。2) FOXM1 (Forkhead box protein M1) 转录因子。FOXM1特异性表达于增殖期细胞中,与细胞生长密切相关,也是Forkhead家族41个因子中唯一在应激条件下表达发生显著变化的因子。抑制FOXM1表达可增加RANKL/OPG比值并促进破骨细胞分化。人牙周膜细胞受压应力刺激后,FOXM1表达降低,促进破骨细胞分化[44]。3) 转录增强缔合域(transcriptional enhanced associatedomain,TEAD) 转录因子。TEAD蛋白是Hippo信号通路的下游效应子,可调节细胞增殖和干细胞功能。机械压应力降低PDLSC中的TEAD1表达,降低OPG表达,从而抑制成骨,促进破骨细胞分化[45]。有研究[46]指出:机械压力可提高牙周膜细胞外泌体中TEAD1的表达,促进巨噬细胞的M1极化,诱发炎症反应。由此可见,TEAD转录因子作为一种新的机械信号响应性转录因子,在牙周膜细胞机械压力信号转导中发挥重要作用。
2.3.4 机械力通过非编码RNA调控破骨细胞的分化 近年来关于非编码RNA调控牙移动及牙周炎机制的研究较多。正畸患者的龈沟液中检测到多种微小RNA的差异表达[47],且非编码RNA在破骨细胞的分化和成熟中具有重要的调控作用[48]。压力作用下, lncRNA FER1L4 表达上调增加了PDLSC中自噬小体的形成和自噬活性的增强,提示lncRNA在正畸治疗中发挥了牙周组织重塑过程中调节自噬机制的作用[49]。还有研究[50]发现:lnc-RNA分化拮抗非蛋白编码RNA (differentiation antagonizingnon-protein coding RNA,DANCR) 在压应力下表达增加,并参与Jagged1诱导的破骨细胞形成及牙根吸收。
2.3.5 机械力诱导PDLSC 释放破骨细胞生成诱导因子调控破骨细胞分化 1) RANKL/OPG比例的改变。机械力作用可通过各种方式激活PDLSC中NF-κB信号通路,并促进RANKL分子的释放,通过该途径调控破骨细胞分化。2) 炎症因子IL和TNF-α等的分泌。促炎反应多数通过这一途径调控破骨细胞分化。3) 气体介质H2S。力学刺激下,PDLSC可释放H2S调控破骨细胞的生成,PDLSC中硫化物的产生有助于其分泌单核细胞趋化蛋白1 (monocyte chemotactic protein 1,MCP1) 和RANKL/OPG系统受体激活剂的表达。这些因子的分泌和表达控制了巨噬细胞的迁移和破骨细胞分化[51]。机械负载刺激的PDLSC产生H2S 通过信号转导与转录激活因子 1 (signal transducerand activatorof transcription 1, STAT1) 信号通路促进巨噬细胞M1极化,这有助于骨重塑和牙移动过程[52]。
2.3.6 机械力诱导PDLSC分泌外泌体蛋白ANXA3促进破骨细胞分化 有研究[53]发现:外泌体作为细胞间交流的重要成分,在压应力作用下促进PDLSC调控破骨细胞分化的过程,但其上游通路仍值得进一步深入研究。
2.3.7 机械力通过调控PDLSC 自噬调控破骨细胞分化 自噬是一种细胞自我保护及稳态维持的机制,通常在细胞中低水平表达维持细胞器稳态。机械压应力刺激增加了PDLSC中自噬蛋白LC3的表达,从而增加了M1巨噬细胞和破骨细胞的数量,以及压力侧牙周膜内M1/M2巨噬细胞的比例[54],间接促进了破骨前体细胞的破骨向分化。机械力作用下PDLSC调控骨骼系统细胞活动的示意图见图1。
3 机械力作用下PDLSC 中调控骨重塑相关的信号通路
3.1 机械力刺激下PDLSC中调控成骨分化信号通路
1)MAPK信号通路。MAPK信号通路是常见的机械敏感相关信号通路,与炎症反应密切相关。MAPK信号通路参与OTM,与破骨细胞的骨吸收和成骨细胞的骨形成有关[55]。机械压力作用通过黏着斑激酶和整合素激活牙周膜细胞内MAPK信号通路[56],MAPK激活可减弱破骨细胞生成,降低OTM患者的TNF-α和NF-κB的表达,促进成骨分化,抑制破骨细胞生成。
2) TGF-β信号通路。TGF-β是细胞外基质重塑和维持组织稳态的关键分子,压应力可抑制PDLSC中TGF-β1和TGF-β3的表达,同时抑制牙周膜胶原的表达[57-58]。有研究[59]指出:抑制TGF-β信号通路可以减少小鼠破骨细胞的数量,增加成骨细胞相关因子的表达。
3) Ca2+- 钙调蛋白依赖性蛋白激酶(Ca2+-calmodulin-dependent protein kinase, CaMK) 通路。CaMK家族包含一系列在骨重塑中起关键作用的蛋白,CaMK信号通路参与成骨细胞分化[60]。基因表达谱的微阵列筛选显示CAMK通路在牙周膜细胞机械反应中具有潜在作用[61]。此外,长期静压力作用可通过CaMK Ⅱ途径增加PDLSC中的OPG表达[62]。
3.2 机械力刺激下PDLSC中调控破骨细胞分化的信号通路
1) NF-κB信号通路。牙周膜细胞中NF-κB配体在静压力作用下显著上调,诱导破骨细胞分化[63]。由PDLSC分泌的RANKL可与核因子-κB受体激活蛋白(receptor activator for nuclear factor‑κB,RANK) 结合,募集肿瘤坏死因子受体相关因子6(tumor necrosis factor receptor-associated factor 6,TRAF6) 并导致下游信号通路(包括NF-κB、c-Fos和MAPK) 激活,是破骨细胞分化的重要介质[64]。
2) AKT信号通路。有研究[54]表明:压力诱导的PDLSC自噬可以通过抑制AKT信号通路促进M1巨噬细胞极化,从而促进炎症骨重塑。
3) αvβ3整合素/ERK信号通路。压力可诱导缺氧相关蛋白骨膜素的表达,通过激活αvβ3整合素-ERK信号通路促进牙周细胞基质金属蛋白酶-2的表达[65]。
4) 糖原合成激酶3β (glycogen synthase kinase-3β,GSK-3β) /β-catenin信号通路。GSK-3β/β-catenin信号通路参与OTM,GSK-3β可减弱Wnt信号通路的作用,抑制骨形成,促进压力侧骨吸收和破骨细胞形成[66] 。
5) Rho激酶信号通路。机械压应力通过Rho激酶途径影响PDLSC中OPN的表达,参与骨吸收和重塑[67]。
3.3 机械力刺激下PDLSC中与成骨分化及破骨分化调控均相关的信号通路
1)Wnt信号通路。Wnt信号通路是胚胎、器官发育的重要信号通路之一。Wnt/β-catenin通路是机械转导的重要组成部分,可调控骨重塑相关基因的转录,被称为成骨细胞分化的中枢调节因子[68]。Wnt5a信号通路参与OTM过程中张力侧的骨形成[69]。此外,Wnt信号通路在破骨细胞分化中也发挥作用,机械力加载1 h后PDLSC的成骨分化显著增强,但12 h后,Wnt/β-catenin通路被激活,RANKL/OPG比值升高,促进了破骨细胞分化[70]。因此,Wnt信号通路的激活既参与成骨也参与破骨调控,具体机制有待进一步研究。
2) Notch信号通路。Notch信号通路交联免疫系统与骨骼系统,在OTM动物模型研究[59]中,压力侧检测到Notch2和Jagged1 (Notch的配体之一)的表达,压力作用上调PDLSC中Jagged1的表达,激活Notch信号通路促进RANKL及IL-6分泌,降低OPG表达,刺激破骨细胞形成。然而也有研究[59]报道:Notch信号可增强PDLSC的成骨分化。这些结果的差异可能是由于细胞施加的力刺激不同所致。
机械力作用下PDLSC中调控骨重塑的应激反应见图2。
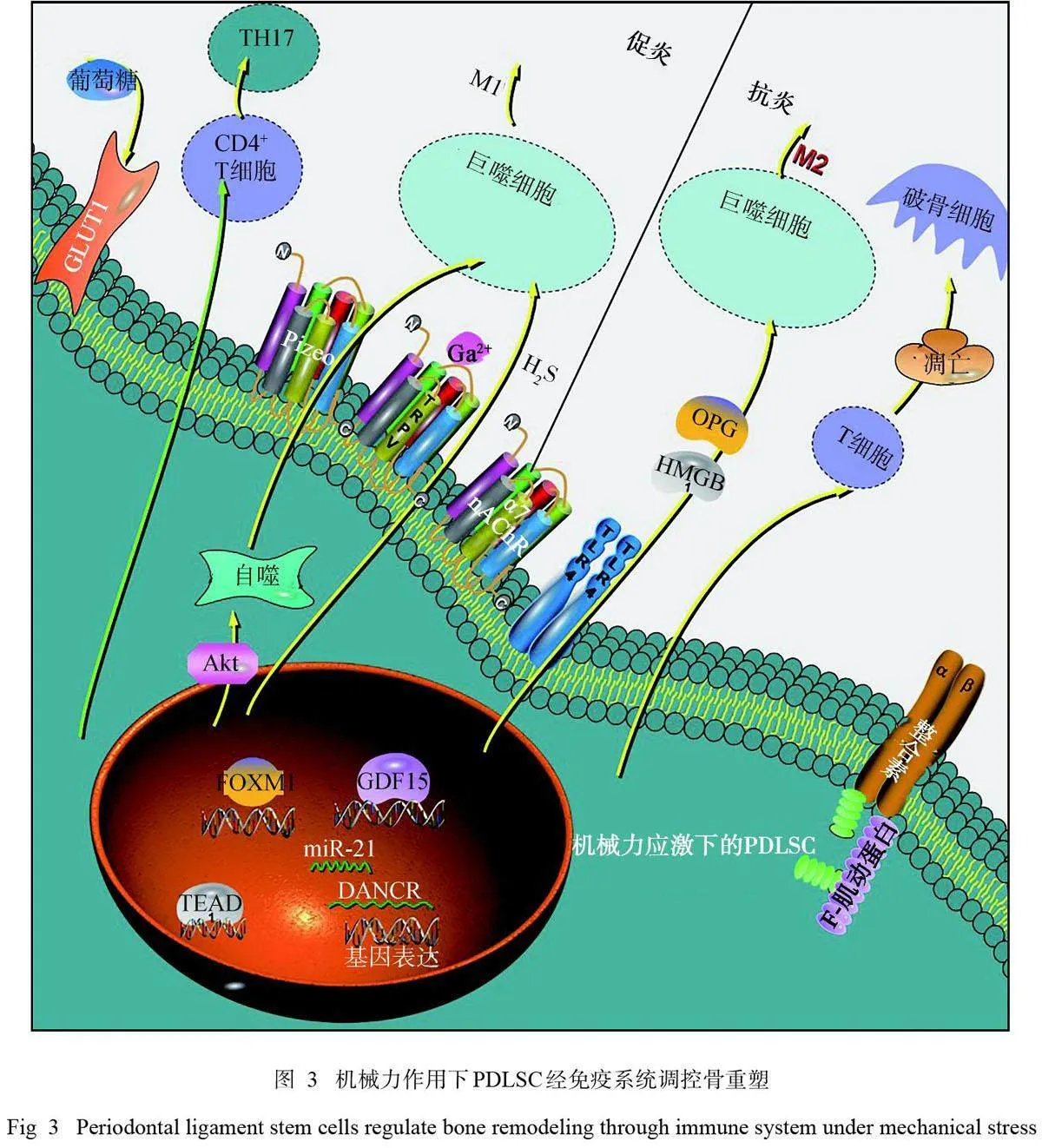
4 免疫调节及神经调节在机械力作用下PDLSC调控骨重塑中发挥的作用
4.1 机械力作用下PDLSC经由免疫相关活动发挥骨重塑作用
PDLSC具有免疫调节的基本特征。OTM是机械力诱导的无菌性炎症,免疫细胞和免疫相关因子均参与力诱导的骨重塑过程,而免疫调节在该过程中发挥重要作用。机械力作用PDLSC可通过免疫细胞间接调控骨重塑过程。一方面,PDLSC可通过分泌炎症因子对正畸力作出反应;另一方面,PDLSC可通过调控淋巴细胞、巨噬细胞的活动发挥骨重塑作用,但目前只有少数研究[71];指出参与其中的细胞包括淋巴细胞、巨噬细胞等,提示骨改建与免疫系统调控密切相关,但缺乏PDLSC在OTM中明确的免疫调节活性研究,其在力学作用下对免疫细胞的直接作用研究证据仍需加强。
4.1.1 促炎作用 PDLSC可对正畸力产生反应,通过分泌各种免疫介质如炎症因子调控骨重塑。机械压应力刺激下PDLSC还可使巨噬细胞M1向极化, 该过程中有气体介质H2S的参与, 并通过STAT1信号通路发挥作用,有助于骨重塑及牙齿移动过程[52]。同时有研究[53] 指出: 力刺激下的PDLSC自噬可诱导巨噬细胞的M1极化,该过程通过AKT信号通路发挥作用,从而促进炎症性骨重塑和OTM过程。还有研究[72]指出:机械压应力刺激下牙周膜细胞中IL-6和TGF-β的表达发生变化,与CD4+T细胞共培养后可促进其Th17向分化,发挥促炎反应,该过程与Notch1信号通路的调控相关。机械力作用下,牙周膜成纤维细胞还可通过释放RANKL激活CD4+T细胞,从而导致OTM过程中的骨吸收[73]。体内研究[74]也表明:OTM过程中,压力侧牙周膜可招募T/B淋巴细胞促进骨吸收。
4.1.2 抗炎作用 在OTM过程中,PDLSC通过释放包括高迁移率族蛋白B1 (high mobility groupprotein,HMGB1) 在内的介质发挥免疫调节潜能,这些介质作为牙周修复的一种途径,改变了巨噬细胞的迁移和分化[75]。机械牵张力作用下,牙周膜成纤维细胞可以产生OPG抑制巨噬细胞的破骨向分化[68]。但关于PDLSC在机械力下通过巨噬细胞实现抗炎的作用有待进一步探索。有研究[76]指出:静压力作用下,PDLSC与T淋巴细胞共培养后,短时间内淋巴细胞凋亡水平增加,破骨细胞活动受抑制;作用12 h后,凋亡水平下降,促进破骨细胞分化。由于PDLSC对机械力的响应取决于机械力的类型及力值大小[77],必须考虑这些因素的影响。
机械力作用下PDLSC经免疫系统调控骨重塑的示意图见图3。
4.2 神经系统参与OTM过程调节骨重塑
近年来,OTM神经调节成为新兴研究热点,研究领域不断扩大。随着神经元引导分子(nervegrowth factor,NGM) 对骨骼发育和骨稳态功能的研究不断深入,已经发现NGM介导的神经系统和骨骼之间存在相互作用[78],神经系统中各种神经肽和受体均参与了骨重塑过程[79]。OTM过程中,牙周膜压力侧有多种神经通路调控PDLSC表面受体及配体的表达。机械压力通过升高细胞内Ca2+浓度上调原代培养的牙周膜细胞中的β2肾上腺素受体(β2 adrenergic receptor,ADRB2) 表达,增加RANKL/OPG比例,促进外周血单核细胞分化为破骨细胞[80]。在OTM过程中,PDLSC作为响应机械力的主要传感器, 其RANKL 和骨硬化蛋白(sclerostin,SOST) 的表达可以被β肾上腺素受体阻断剂所抑制,表明交感神经系统(sympatheticnervous system, SNS) 可以调控PDLSC 影响OTM[79]。还有研究[81]表明:感觉神经标志物P物质(substance P,SP) 具有上调牙周膜细胞中RANKL和血红素加氧酶-1表达并下调OPG表达的作用。综上,神经系统通过多种途径参与调节成骨、破骨细胞分化以及OTM期间牙周膜细胞的代谢,包括中枢和外周循环中神经肽的作用、神经源性无菌性炎症的介导以及牙周膜细胞表面受体表达的调节。然而,神经肽和神经系统如何诱导牙周膜炎症并因此影响牙槽骨重建的详细机制仍不清楚,进一步的探索有助于更深入地理解OTM中神经调节的机制,调控加速OTM进程[82]。
5 细菌性炎症微环境对机械力作用下PDLSC调控骨重塑的影响
牙周炎是牙周组织进行性破坏的慢性感染性疾病,细菌感染及伴随的宿主免疫反应形成独特的微环境,对牙周膜中驻留的PDLSC产生影响。炎症微环境中,PDLSC的分化和再生能力受到抑制。炎症条件下,PDLSC中组蛋白去乙酰化酶抑制剂(histone deacetylase inhibitor, HDAC9) 会降低PDLSC在炎症条件下的成骨分化能力,具体机制表现为HDAC9和miR-17结合形成一个抑制环,对miR-17的抑制加重了炎症环境中PDLSC钙化结节的丢失,并阻断了HDAC抑制剂在挽救成骨中发挥的作用[83]。牙周炎微环境会造成PDLSC生物学功能的损伤和力学敏感性异常,有研究[84]认为这种差异与m6A甲基转移酶修饰相关。机械力刺激下,炎症微环境来源的PDLSC中m6A修饰存在差异,可以通过靶向JAK1信号通路调节间充质干细胞的多向分化潜能[84]。炎症来源的PDLSC对静态牵张力更加敏感,炎症PDLSC的增殖及成骨分化减弱,表观遗传调控在这一过程中发挥重要调控作用[85]。牙周炎患者的PDLSC表现出较低的免疫抑制活性[86],因此探讨PDLSC在OTM期间的免疫调节行为,可能会进一步阐明OTM与牙周炎的相互作用。应激条件下,如炎症微环境,可以通过上调PDLSC细胞内自噬水平促进成血管因子的表达,促进血管再生[87],因此探索炎症微环境下PDLSC对机械刺激的反应可为牙周疾病的防治提供一定的研究基础。
6"结语
综上所述,机械力作用下PDLSC一方面直接调控骨骼系统的细胞活动调控骨重塑,另一方面通过调控免疫活动间接调控骨重塑,神经调节也参与了机械力激活PDLSC的过程。但现存研究尚有许多不足:就研究模型而言,PDLSC的体外机械力加载模型尚未建立标准;就PDLSC感知机械刺激而言,仍有许多机械敏感受体的下游通路尚不清楚;就PDLSC调控骨重塑而言,PDLSC经同一基因及相同信号通路对骨重塑的调控存在差异。以上问题都需要更多的研究来探讨,但目前阶段的研究仍揭示出神经调节及非编码RNA在PDLSC感知机械刺激中发挥的重要作用,以及免疫系统活动在PDLSC调控骨重塑中的作用,这为机械力诱导骨重塑的研究提供了一定基础。
利益冲突声明:作者声明本文无利益冲突。
