
利用CRISPR/Cas9基因编辑片段敲除技术创制玉米多样化等位变异
2024-10-16 00:00:00张茂林王娟严佳丽何春梅徐倩刘铁山董瑞刘春晓关海英刘强汪黎明曾廷儒
山东农业科学 2024年9期

摘要:利用CRISPR/Cas9基因编辑技术创制等位变异已经成为众多植物中增加种质资源多样性的重要手段。提高玉米等谷物中类胡萝卜素含量是发展中国家解决维生素A缺乏症的一种经济方法。本研究以玉米中调控类胡萝卜素合成的基因crtRB1为研究对象,利用双靶标片段缺失性基因编辑技术,在原生质体中对crtRBI的启动子顺式元件进行片段敲除,获得了具有片段缺失和InDel(插入或缺失)的多样化等位变异,这为在玉米中实现稳定的crtRB1启动子元件的缺失突变奠定了基础,对启动子元件的功能研究和玉米的品质改良具有重要意义。
关键词:玉米;crtRBI;基因编辑;片段敲除;启动子
中图分类号:Q781:S503.531+3 文献标识号:A 文章编号:1001-4942(2024)09-0001-05
粮食安全是世界首要问题,随着人口不断增长,现有的粮食产量和品质越来越不能满足人们的需求。玉米是三大作物之一,是我国乃至世界种植面积最大、产量最高的作物。玉米不仅是粮饲兼用作物,也是能源产品的重要原料植物。玉米传统育种近几十年取得长足的进步,但主要基于现存的自然变异进行人工选择,再通过杂交聚合选育新品种,费时费力。然而,现存的变异远远不能满足现代育种的需求,这就需要用生物技术手段人工创制遗传变异新种质,以缩短育种周期,提高育种效率。
近年来,基因编辑技术的研究和发展突飞猛进,新兴的CRISPR/Cas9基因编辑技术为育种家提供了能够精确创造等位基因的大好机遇。CRISPR/Cas9基因组编辑技术最早于2010年在原核免疫系统中被发现,也是继第一代锌指结构核酸酶(ZFNs)和第二代转录激活子样效应因子核酸酶技术(TALENs)之后,被公认的第三代基因组编辑技术。植物主要通过非同源末端连接(NHEJ)途径来修复双链DNA断裂缺口(DSBs),以此进行碱基的敲除和敲入,此过程可以导致基因移码突变,使目的基因功能缺失,从而达到敲除基因的目的。
CRISPR/Cas9基因组编辑技术在2013年第一次应用于植物中,之后便成为植物科学界的明星技术被广泛应用,如拟南芥、玉米、水稻、大豆、番茄等。至此,CRISPR/Cas9在农作物中的应用已非常流行,近几年用在农作物上创造了很多有益变种,涉及产量、品质、抗病和抗逆等。CRISPR/Cas9技术操作简单,编辑效率高,可用来创造多倍体植物的突变体、基因组大片段的敲除以及产生多基因突变体等。当同一条染色体相距一定距离的两个靶位点同时被编辑时,靶标之间的片段有一定的几率被敲除。在模式植物拟南芥中,Zhao等通过此方法将含有多个基因的基因组大片段成功敲除:另外,水稻中就有长达245 kb的基因组片段用此方法被剪切掉。Rodriguez-Leal等利用CRISPR/Cas9基因组编辑技术对SICLV3启动子顺式元件进行片段敲除,创造了多样化的等位变异,并筛选得到了具有连续表型的多样化顺式元件突变体,并可以直接作为育种的种质资源,为分子育种提供了新的思路和方法,使得通过改造玉米启动子获得理想的变异新种质成为可能。这些等位突变如果用传统的方法需要花费大量的时间和精力去筛选,而这种基于CRISPR/Cas9基因组编辑技术的启动子变异方法成功绕开了传统育种的限制,直接创造并且筛选获得了科学家渴望得到的变异,为突破分子育种瓶颈提供了新的思路。
crtRBI是调控玉米籽粒中维生素A含量的关键基因,该基因中一个稀有变异降低了基因的转录水平,大幅提高了玉米籽粒中维生素A的含量。此举惠及患有维生素A缺乏症的数万非洲儿童。本研究利用CRISPR/Cas9基因编辑技术对玉米crtRB1基因的启动子区域进行编辑,旨在进一步明确crtRBI的功能,同时为有效调节玉米籽粒中维生素A的含量提供理论参考。
1材料与方法
1.1试验材料
本研究所用植物CRISPR/Cas9基因编辑载体pCPB-ZmUbi-hspCas9由谢传晓研究员提供。DL2000 DNA Marker、T4连接酶、DNALoading Buffer、DNA Marker、FastPfu、2x Taq Mix、大肠杆菌DH5a、EHA105农杆菌感受态细胞等购自北京全式金生物技术有限公司:限制性内切酶BsaI购自纽英伦生物技术(北京)有限公司;高保真酶购于南京诺唯赞生物科技股份有限公司:氯仿,无水乙醇、异丙醇等试剂均购于国药集团。引物由青岛擎科梓熙生物技术有限公司合成,基因测序由上海生工生物工程技术服务有限公司完成。
1.2双靶位点设计
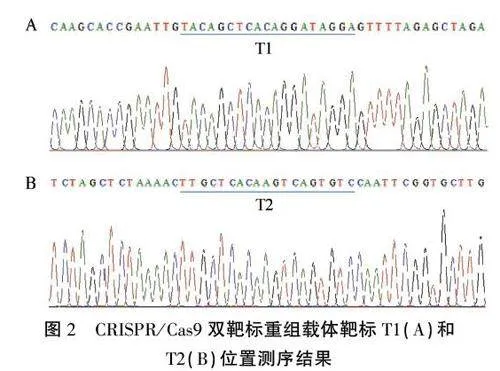
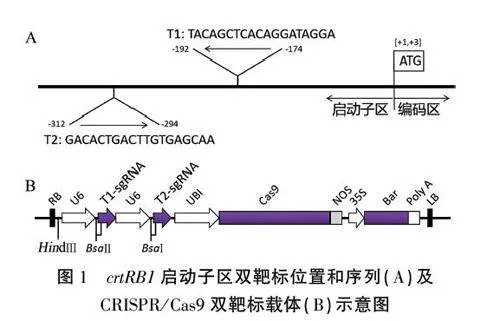
由于转化所用受体自交系KN5585的基因组和B73的基因组有一定的差异,我们首先以B73基因组做参考,对选取的需要进行片段敲除的序列两端设计引物PRB/-T-F和pRBl-T-R(表1),然后以KN5585的DNA为模板扩增测序确定要进行片段敲除的启动子区域序列。通过CRISPR-P 2.0在线设计网站(http://crispr.hzau.edu.cn/cgi-bin/CRISPR2/CRISPR)设计crtRBI启动子目标区域的2个靶位点Tl和T2(图1A)。
1.3片段敲除载体构建
1.3.1靶标引物退火形成双链片段 靶标T1对应的两个引物是pRBI-T1-F和PRB/-T1-R,靶标T2对应的两个引物是pRB/-T2-F和pRB1-T2-R,将每个靶标对应的两条引物分别成对混合,直接退火形成双链:37℃20 min,95℃5min,以6℃/min的速度降至25℃,形成带粘性末端Bsa I酶切位点的靶标片段,备用。
1.3.2sgfRNA-U6片段的获得 以pCPB-ZmUbi-hspCas9质粒为模板,用引物sgU6Fl/sgRNA-R扩增片段sgRNA,用U6-F/sgU6R1(表1)扩增U6片段,然后将两个PCR产物混合后为模板,利用两个片段之间的重叠序列,以引物sgU6F1/sgU6R1(表1)(带BsaI酶切位点)进行重叠PCR。反应体系:上、下游引物(10 umol/L)各2uL,2x Buffer 25 uL,dNTP Mix(10 mmol/L)1uL,两个模板产物均为1 uL,PhantaMax高保真DNA聚合酶1uL,用ddH2O补至50 uL。反应程序:95℃1min;95℃15 s、55℃15 s,72℃30 s,35个循环;72℃5min。电泳,选择合适大小的条带切胶回收,回收产物用BsaI酶切纯化,获得带BsaI粘性末端的sgRNA-U6片段,备用。
1.3.3载体线性化 载体pCPB-ZmUbi-hspCas9中具有毒性致死基因ccdb,使用限制性内切酶BsaI将载体中的ccdb切除,形成两端具有BsaI粘性末端的线性化载体,备用。
1.3.4连接形成最终载体 构建敲除载体的每个靶位点分别由独立的U6启动子驱动(图1B)。将上述获得的T1和T2靶标片段、sgRNA-U6片段、线性化载体pCPB-ZmUbi-hspCas9用T4连接酶进行链接组装,体系为:3个片段各0.5 uL,T4连接酶0.3 uL,加水至5uL。25℃连接1h。将连接产物转化大肠杆菌DH5a,之后于LB平板培养基(含Kan)上培养,挑取单克隆用引物BGK-T-F/BGK-T-R(表1)进行菌落PCR鉴定及测序,比对筛选正确的克隆提取质粒。
1.4玉米原生质体的提取和转化
参考文献中玉米原生质体的制备及转化方法并稍作调整。将玉米种子在花盆中培养(25℃,16L:8D)8 d左右,取幼苗叶中间段切成宽0.5-1.0 mm的细条,转移到酶解液[含0.6mol/L甘露醇、10 mmol/L MES(pH值5.7)、1.5%纤维素酶R10、0.75%离析酶、10 mmol/L CaCl2.0.1% BSA];用锡箔纸包住后放置28℃摇床,50r/rmn避光酶解5 h:加入等体积W5(含154mmol/L NaCI、125 mmol/L CaCL2、5 mmol/L KCl、2mmol/L MES),轻柔摇晃均匀后过40 um细胞筛:加入MMG溶液(含0.4 mol/L甘露醇、15mmol/L MgCl2、4 mmol/L MES)重悬原生质体;将构建好的质粒利用PEG介导的方法转入原生质体,避光条件下28℃培养48 h:离心收集玉米原生质体,用DNA快速提取试剂盒提取基因组DNA。
1.5基因编辑方式检测
以提取的原生质体DNA为模板,使用特异性引物pRB1-T-F/pRBl-T-R进行PCR扩增,将PCR产物纯化后连接pEASY-Blunt载体,并将连接产物转化大肠杆菌DH5a,涂板LB固体培养基(Kana),37℃倒置培养过夜。等菌落长到合适大小后,选取大于50个独立的菌落分别进行PCR扩增,将扩增产物送至上海生工生物工程技术服务有限公司测序。通过测序结果比对判定两个靶位点之间有无大片段缺失以及两个靶位点各自区域的编辑情况(插入、替换或缺失)。
2结果与分析
2.1片段敲除载体的构建及鉴定
以构建好的片段敲除载体质粒为模板,PCR扩增后分别用上游和下游引物测序,通过比对测序结果中靶标T1(BGK-T-F测序)和T2(BGK-T-R测序:序列反向互补)以及侧翼序列,筛选获得了预期的重组载体(图1B,图2),证明所需的片段敲除载体构建成功。
2.2原生质体转化及编辑情况检测
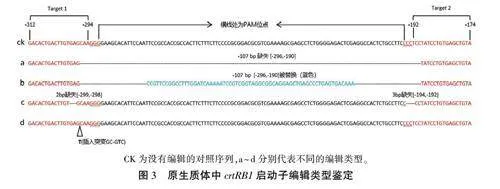
对原生质体DNA进行PCR扩增,产物连接pEASY-Blunt载体后转化大肠杆菌DH5a,长出菌点后,选取独立的50个进行菌落PCR扩增并测序。通过对测序结果进行比对(图3)发现,目标编辑区域发生了大片段缺失和替换,同时,两个靶标各自位点也发生InDel(替换、插入或缺失)。发生编辑的位点大部分是双等位基因,只有6%(50个中有3个)包含一个野生型拷贝。双靶标之间发生大片段缺失的概率为8%(图3a),发生大片段替换的概率为2%(50个中有1个)(图3b)。两个靶标各自位点也有InDel,T1位点发生的编辑类型包括2 bp缺失(图3e)和单碱基(T)插入(图3d),概率分别为48%和16%;T2位点发生了3 bp的缺失(图3e),概率为32%。对于单个突变体来讲,两个靶标位点编辑方式都有InDel的概率为8%(图3e),只有一个靶标位点区域有InDel的概率为8%(图3d)。表明本研究所用的CRISPR/Cas9基因编辑系统编辑效率较高,能够满足片段敲除和多样化等位变异的需求。
3讨论与结论
在育种过程中,经常遇到需要微调某种性状来寻找众多育种性状的最优平衡点,从而达到品种选育与改良的目的。基因编辑技术的突飞猛进给育种带来了重大机遇。利用CRSIPR/Cas9双靶标基因编辑技术,对启动子的顺式作用元件进行片段缺失性突变,来调节基因的功能,能够在创制新的等位突变的同时,微调相应性状,从中选择最优的新种质资源应用到育种中,从而达到品种改良的目的。
本研究将构建的CRSIPR/Cas9双靶标片段敲除载体转入玉米原生质体,通过编辑方式和效率检测,证明该系统的有效性,能够满足片段敲除的需求,同时编辑方式的多样性具备创制多样化变异材料的条件。本研究结果可为评估启动子顺式元件的功能及其在育种中的价值提供理论参考,也为发展一套全面详细研究启动子顺式元件的方法以及评估启动子不同调控元件在育种中的应用潜力提供理论支撑。至此,通过人工改造顺式元件来调控基因功能成为可能,作物遗传改良已经进入一个新的时代,需要我们共同探索。
